
Our group focuses on the development of a novel rearrangement we call the 1,3-diaza-Claisen rearrangement. The reaction involves the reversible addition of a tertiary, allylic amine to a heterocumulene such as a carbodiimide, isocyanate or isothiocyanate to afford a zwitterionic intermediate. The zwitterionic intermediate then undergoes a rate-determining 3,3-gimatropic rearrangement to afford a guanidine, urea or thiourea product depending on the heterocumulene.
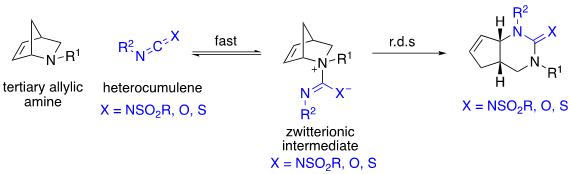
We have additionally developed tethered variants of the reaction in which an in situ generated carbodiimide, tethered to a tertiary allylic amine reacts intramolecularly to form the zwitterionic intermediate that in turn undergoes a 1,3-diaza-Claisen rearrangement to afford a tricyclic guanidine in a single step.
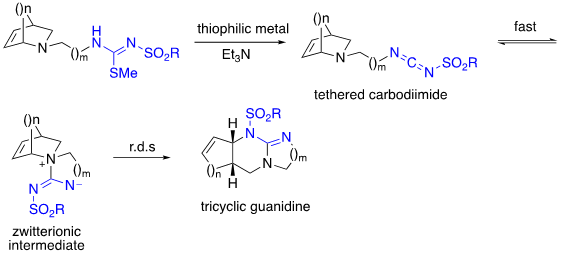
While the 1,3-diaza-Claisen rearrangement has worked well with bridged-bicyclic tertiary allylic amines, for the reaction to be widely applicable, it must also work with simpler tertiary allylic amines which we have found to be more challenging. To address this challenge, we have deployed the tethering strategy in which a simpler tertiary allylic amine tethered to in situ generated carbodiimide forms a zwitterionic intermediate intramolecularly. Unlike with the bridged bicyclics these do not readily undergo rearrangement at room temperature and are thus isolable. They readily rearrange when heated in an appropriate solvent through a chair-like transition state as determined through DFT calculations to afford the product.
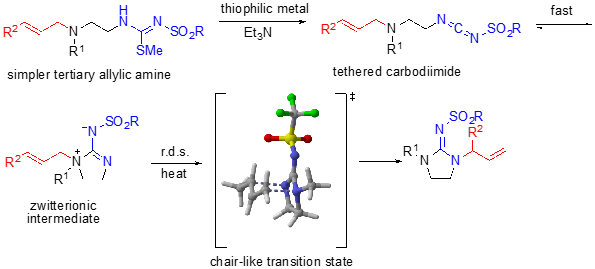
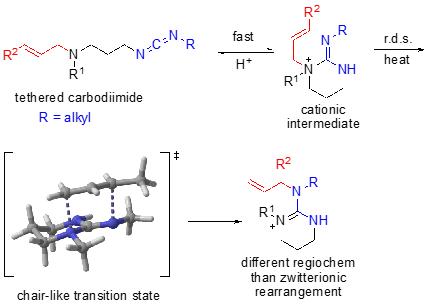
We have additionally developed a cationic, tethered 1,3-diaza-Claisen rearrangement in which a carbodiimide in the presence of acid affords a cationic intermediate, rather than a zwitterionic intermediate. In this scenario, DFT calculations show that protonation occurs regioselectively such that rearrangement occurs on to the exocyclic nitrogen affording the alternate regiochemistry to the zwitterionic rearrangement.
In pushing the applicability of the 1,3-diaza-Claisen rearrangement, we have also explored its application toward ring expanding reactions of vinyl pyrrolidines tethered to carbodiimides. DFT calculations have again shown that for these reactions, the cationic rearrangement pathway is a lower energy pathway than the zwitterionic rearrangement pathway.
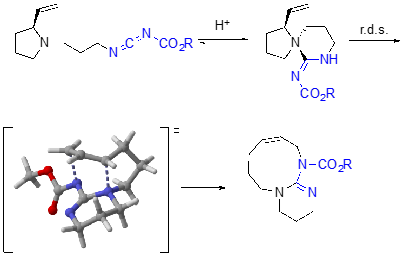
Future directions include the development of diastereoselective 1,3-diaza-Claisen rearrangements, chirality transfer 1,3-diaza-Claisen rearrangements, catalytic and asymmetric catalytic 1,3-diaza-Claisen rearrangements as well as the application of the 1,3-diaza-Claisen rearrangement to the synthesis of guanidine natural products. DFT calculations have been transformative in understanding the project as well as elucidating reactivity trends, and will continue to be part of our tool box for moving the project forward. Finally, we are also working on a different sigmatropic rearrangement that will afford carbodiimides.
