
Pomeroy Emeritus Professor — Analytical Chemistry, Mass Spectrometry, Proteomics, Metabolomics
Research and/or Creative Works
My laboratory uses mass spectrometry to measure biological compounds.
A primary focus is measurement of metabolic pathways in vivo and in cell culture, something my laboratory has been doing for 40 years, although this work is now called “metabolomics”. In most cases we label metabolites with nonradioactive stable isotopes to increase their mass and be measurable and differentiable by mass spectrometry from endogenous compounds already in humans, animals, tissues or cells. We develop kinetic models to measure rates of biochemical processes. Many of these measurements have been made directly in humans.
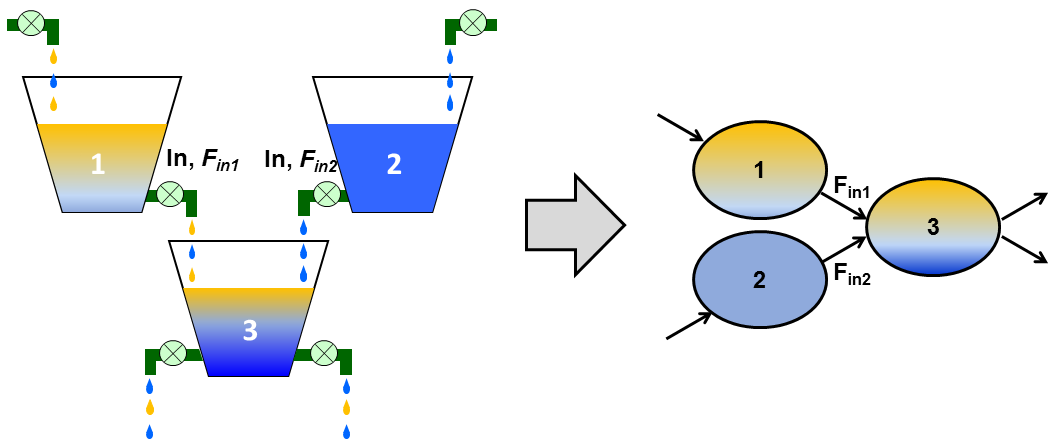
The figure below shows our “bucket” model approach to kinetics. The buckets show water dripping from #1 and #2 into #3. Yellow dye (the stable isotope label) is added to bucket #1 and the amount of dye determined in bucket #3 – that allows determination of how much water enters from bucket #1 into #3. The same applies on the right where now measure rates of biochemical metabolites being converted.
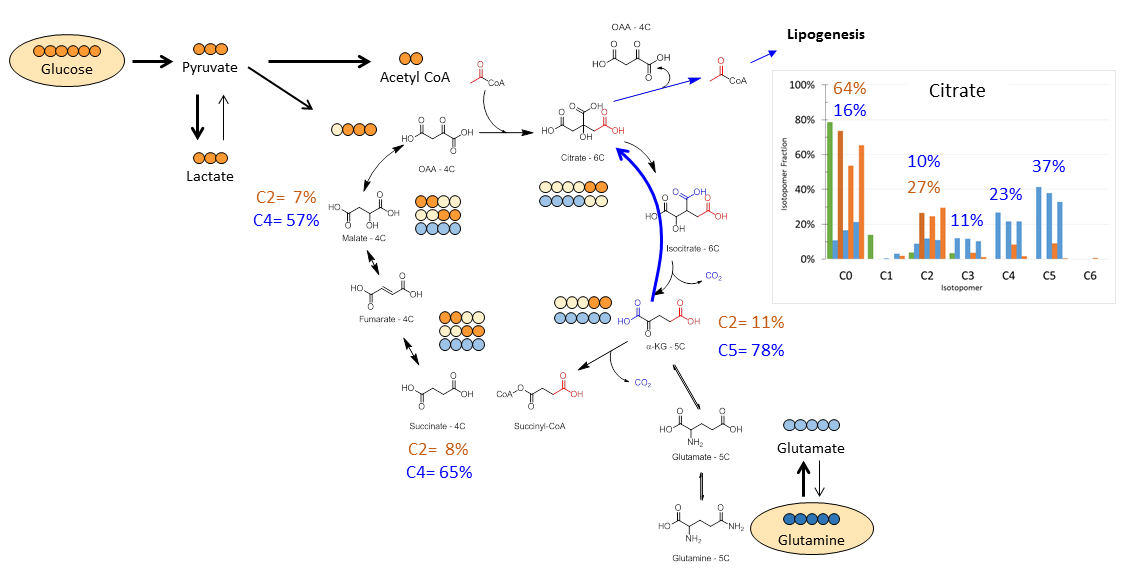
This scheme can be used to measure many metabolic pathways. The example below shows different amounts of 13C label in citrate in the TCA-cycle when cells were given 13C-glucose versus 13C-glutamine. That rates of glycolysis, oxidative phosphorylation and production of fat can be determined from these substrates (glucose and glutamine).
We also use mass spectrometry to measure peptides and proteins in the area of proteomics. Proteins have many functions from being the basic unit of our muscles to enzymes to being key signaling molecules. The regulation of all of these functions is complex and often involves posttranslational modification (PTM) of proteins through phosphorylation and other covalent changes. An area of research has been development of proteomic methods not for identifying, but for quantifying PTMs. The figure below from M.J. Previs et al., Anal. Chem. highlights our first scheme to quantify phosphorylation of proteins at multiple sites using a tryptic digest of proteins and LCMS analysis. The key to the method is to perform quantification of phosphorylation without having to measure the phosphorylated peptide per se. This approach measures the phosphorylation (PO4) site peptide X against one or more “reference” peptides Z before (2) and after the sample has been dephosphorylated (2a).
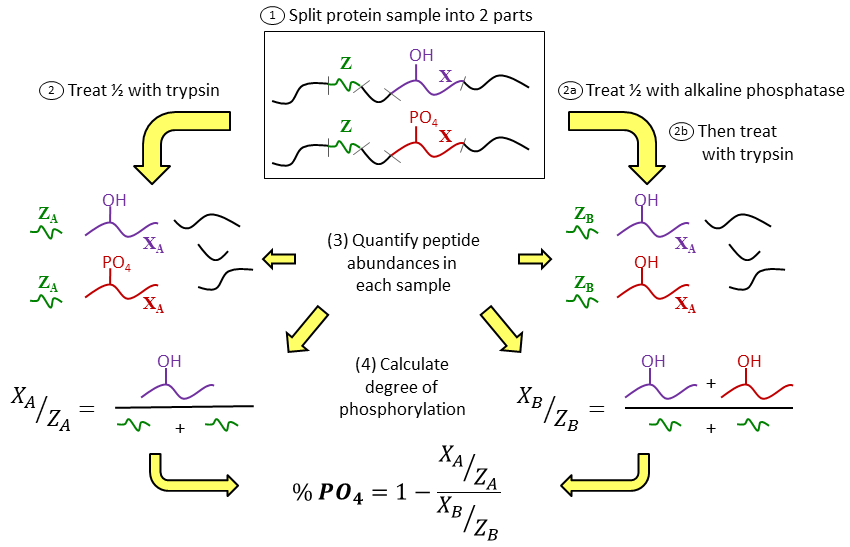
We have since improved on this scheme by using recombinant proteins that contain no PO4 to replace the dephosphorylation step.
Publications
For complete lists of publications go to NIH NLM MyNCBI and Google Scholar
- JR Brooklyn & DE Matthews. Simple, rapid spectrophotometric assay of dispensed methadone for diversion control. J. Addict. Med. 16: 671-677, 2022.
- KS Sidhu, E Amiel, RC Budd & DE Matthews. Determination of cell volume as an integral part of metabolomics. Am. J. Physiol. Cell Physiol. 321: C947-C953, 2021.
- CHP van den Akker, JB van Goudoever & DE Matthews. General concepts of protein metabolism. In: Fetal and Neonatal Physiology, RA Polin, SH Abman, DH Rowitch & WE Benitz, eds. 6th ed, vol. 1. Philadelphia, PA: Elsevier, 2022, 400-407.
- EL Giddings, DP Champagne, M-H Wu, JM Laffin, TM Thornton, F Valenca-Pereira, R Culp-Hill, KA Fortner, N Romero, J East, P Cao, H Arias-Pulido, KS Sidhu, B Silverstrim, Y Kam, S Kelley, M Pereira, SE Bates, JY Bunn, SN Fiering, DE Matthews, RW Robey, D Stich, A D’Alessandro & M Rincon. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 12: 2804, 2021.
- ME Jennings, JR Silveira, KM Treier, PB Tracy & DE Matthews. Total retention liquid chromatography–mass spectrometry to achieve maximum protein sequence coverage. Anal. Chem. 93: 5054-5060, 2021.
- DE Matthews. Review of lysine metabolism with a focus on humans. J. Nutr. 150: 2548S-2555S, 2020.
Awards and Recognition
- 2014 — Pomeroy Professor of Chemistry, University of Vermont
- 2007 — Member, Vermont Academy of Science and Engineering
- 2004 — University of Vermont University Scholar

Areas of Expertise and/or Research
Analytical chemistry, mass spectrometry, proteomics, metabolomics, stable isotope tracers, and in vivo metabolite flux and kinetic measurements
Education
- B.A. — DePauw University, Greencastle, Indiana, May 1973.
- Ph.D. — Analytical Chemistry, Indiana University, Bloomington, Indiana, December 1977.
Contact
- (802) 656-8114
Innovation Hal E339