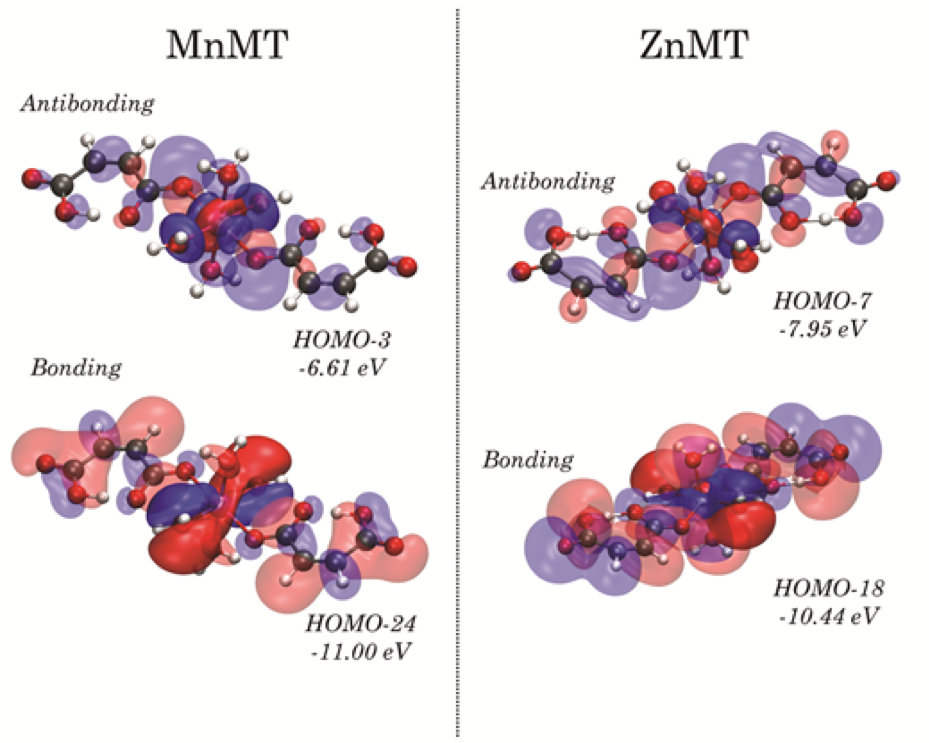
Density functional theory (DFT) is one of the most utilized computational tools in chemistry, physics, and materials science. Based on quantum mechanics, DFT methods enable highly-accurate calculation of atomic and molecular properties, and is able to lend significant insight into the fundamental origins of many bulk physical properties. Using DFT methods, research in the Ruggiero Lab focuses on understanding the origins of atomic and molecular dynamics, and how such dynamics influence the properties of materials on the bulk scale. In addition to using computational methods, we also collaborate heavily with the developers of the CRYSTAL software package based in Turin, Italy, and are regularly helping to develop new algorithms to support our experimental work.