Policies and Procedures for the Institutional Biosafety Committee
Search for a Word
To search for a word use the keyboard shortcuts CTRL-F or F3. Mac users should use Apple's Command key (⌘) + F.
Section I: Committee Information
Section II: Research Guidance
- 2.0 Operations of the IBC
- 3.0 Master Protocol Registration (MPR) Submission
- 3.1 Initial PI Risk Assessment
- 3.2 Risk Assessment
- 3.3 Research Including Recombinant DNA or Synthetic Nucleic Acid Molecules
- 3.4 Research Including Animals
- 3.5 Research Including Infectious Agents
- 3.6 Research Including Biotoxins
- 3.7 Research Including Select Agents and Toxins
- 3.8 Research Including Human Subjects
- 3.9 Projects Involving Plants (NIH Appendix P)
- 3.10 Dual Use Research of Concern
- 4.0 Subsequent Submission Requirements
- 4.1 Amendment to Approved Master Protocol Registration
- 4.2 Addition of a New Funded Project to an Approved Master Protocol Registration
- 4.3 Continuing Review of Master Protocol Registration
- 4.4 Laboratory Accidents/Exposures/Spills (incidents)
- 4.5 Closing a Master Protocol Registration by PI
- 4.6 Closure of a Registration or Amendment by Committee
- 5.0 Investigator Responsibilities
- 6.0 Conflict of Interest
- 7.0 Post Approval Monitoring
- 8.0 Procedures for Managing Noncompliance
- 9.0 Animal/Biosafety Level 3 Facility
Section I
1.0 Mission Statement
The University of Vermont (UVM) Institutional Biosafety Committee (IBC) is a standing committee that ensures that all research and teaching activities involving biohazardous materials are conducted in a safe and informed manner. The IBC is responsible for ensuring full compliance with the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines) and for monitoring all other research and teaching activities involving the use of infectious or potentially infectious biological materials and biotoxins. The IBC reviews all research and teaching protocols that involve the following materials without regard to the source of funding:
Recombinant or synthetic nucleic acid molecules as specified in the NIH Guidelines
Human, animal, and plant pathogens (bacteria, fungi, viruses, parasites, prions)
Plasmid vectors
Viral vectors
Human-derived materials (blood, blood products, cells, tissues, and clinical specimens) when used in conjunction with recombinant or synthetic nucleic acid molecules
Biotoxins
Select Agents and Toxins
The materials listed above will hereafter be referred to in this document as “biohazardous materials” or “materials”.
This document outlines the policies and procedures that must be followed when using or storing biohazardous materials. Since laboratory work can involve exposure not only to these biohazardous materials, but also to other biohazards, chemical and radiological hazards, these policies should be used in conjunction with other pertinent University policies. See Radiation Safety Committee and UVM’s Environmental Health and Safety Committee web pages for more information.
Revised: 07/24/2024
1.1 Introduction to the Committees
The Institutional Biosafety Committee (IBC) at the University of Vermont (UVM) serves the UVM community. The IBC is responsible for review of research projects and teaching-related activities to ensure that recombinant or synthetic nucleic acid molecules, infectious agents and biological toxins are handled appropriately at UVM.
The IBC Policy and Procedure Committee, which includes Committee leadership, Biosafety Professionals, and IBC staff, convenes monthly to review changes in policy and procedures and new regulations.
Ad-hoc Noncompliance Subcommittees, including a subset of the members and other institutional personnel as applicable, are convened as necessary to review noncompliance cases.
Institutional Review Entity (IRE) is convened when a project appears to be Dual Use Research of Concern as outlined in Section 3.10 of this document. This review body consists of various constituents across campus as outlined in Section 3.10.
Revised 03/13/25
1.2 Governing Principles
The NIH Office of Science Policy is where the IBC turns when developing local procedures and policies. Two primary resources are listed below:
NIH Guidelines Web Page
This page provides up-to-date NIH news, register notices, director’s statements, etc. This is also where the latest NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines) is found. The Guide requires that each institution establish an Institutional Biosafety Committee with the authority to approve proposed research projects and teaching activities involving recombinant or synthetic nucleic acid molecules. The Guide details safety practices and containment procedures for basic and clinical research involving recombinant or synthetic nucleic acid molecules, including the creation and use of organisms and viruses containing recombinant or synthetic nucleic acid molecules.
Centers for Disease Control and Prevention (CDC)
The CDC in collaboration with NIH published the Biosafety in Microbiological and Biomedical Laboratories (BMBL), 6th Edition. This document contains guidelines for microbiological practices, safety equipment, and facilities that constitute the four established biosafety levels. The BMBL is generally considered the standard for biosafety.
Revised: 07/24/2024
1.3 Scope of Authority
The President of the University has delegated the authority to the Vice President for Research Administration as the lead Institutional Official responsible for the assurance of compliance in the biosafety area. The IBC policies apply to activities involving the use of biohazardous materials in research projects and applicable teaching laboratories. It applies to research and teaching activities that are:
Sponsored by UVM,
Conducted by UVM researchers,
Classes taught by UVM faculty, or
Conducted using UVM’s property, facilities, or non-public information.
All faculty, staff, students, and visitors are included within the scope of these IBC Policies.
The IBC is delegated the authority to:
Define the basic policies, procedures and standards as required by NIH to oversee the safe use of biohazardous materials.
Review requests for the use of these materials for compliance with NIH Guidelines and the BMBL, and approve those requests which are found to conform. As part of the review process, the IBC will do the following, as applicable:
Conduct an independent assessment of the containment levels, as required by the NIH Guidelines for research involving recombinant or synthetic nucleic acid molecules.
Conduct an assessment, if applicable, of the facilities, procedures, practices, training, and expertise of personnel involved in the requested use of these materials.
Ensure compliance with all surveillance, data reporting, and adverse event reporting requirements set forth in the NIH Guidelines.
Disapprove, terminate, or suspend activities involving these materials which are not in conformity with the Guidelines;
Notify investigators in writing of its decision to approve or withhold approval of activities involving these materials, or of modifications required to secure IBC approval. All decisions will be part of the IBC records maintained by the Research Protections Office;
Set containment levels as specified in the NIH Guidelines;
Conduct periodic review of the use of these materials to ensure that the requirements of the Guidelines are being fulfilled;
Consult with the biosafety team in maintaining and following emergency plans covering accidental spill and personnel contamination resulting from use of recombinant DNA, infectious agents or biological toxins;
Report to the Vice President for Research Administration any significant related illness or accident resulting from use of these materials that appears to be a hazard to public health;
Report to the Vice President for Research Administration and the Office of Science Policy (OSP) any significant problems with or violation of the Guidelines;
The IBC may not authorize initiation of experiments not explicitly covered by the Guidelines until NIH establishes the containment requirement.
Institutional Relationship
The following organizational chart outlines the direct lines of responsibility and corresponding authority.
University of Vermont and State Agricultural College Board of Trustees |
President (CEO) |
Provost |
Vice President for Research Administration Authorized Institutional Official (IO) |
Research Protections Office (RPO) |
Institutional Biosafety Committee (IBC) |
Revised: 07/29/2024
1.4 Committee Membership
Committee membership is comprised of members with varying professional and personnel backgrounds who have demonstrated a genuine interest in and commitment to the purpose of the Committee.
Committee leadership and members serve at the discretion of the Institutional Official (IO.) The IO has delegated signature authority to the Executive Director of Research for the appointment letters.
Chair
- Committee Chairs are appointed by the IO
- A Committee Chair must be a University faculty Member and must have prior service as a Committee Member.
- It is the responsibility of the Committee Chair to conduct Committee meetings in accordance with established federal regulations and University operating policies and procedures. These responsibilities, include but are not limited to the following:
- sign official Committee action documents
- keep abreast of relevant state and federal regulations;
- meet as needed with the IBC Director and/or IO to discuss Committee activities;
- meet regularly with appropriate representatives of the RPO and the Environmental Health and Safety Office to coordinate the review of biohazardous materials throughout the University;
- recommend, in consultation with the IBC Director, new Members to the IO;
- ensure that new Members are properly oriented to and educated about their duties and responsibilities;
- initiate activities designed to keep the campus and community apprised of IBC activities; and
- assist appropriate University administrators in the preparation of federal reports and assurances and meet with inspectors/site visitors as necessary.
- No method for removal is delineated, as all Members are appointed and serve at the discretion of the IO.
- The Chair is appointed for renewable two-year terms.
Associate Chair
An Associate Chair is nominated by the Chair from among Membership of the IBC. Responsibilities of the Associate Chair are to
- conduct the meetings of the IBC in the absence of the Chair or if there is a conflict of interest by the Chair's participation,
- sign official Committee action documents and approve Committee actions as delegated by the Members or by designation of the Chair;
The Associate Chair is appointed for renewable two-year terms.
Regular Members
Membership will comply with all relevant federal and state regulations. Every effort will be made to consider gender and cultural diversity.
- The IO appoints all Committee Members, after receiving recommendations from the IBC Chair and the IBC Director.
- When vacancies occur, suggestions for Membership will be sought, after which formal recommendation(s) for new Member(s) will be made by the Committee Chair and the IBC Director to the IO.
- The IO will in no case make a final appointment without prior consultation with the Committee Chair and the IBC Director.
- Committee appointments will usually be for renewable two-year terms, serving at the discretion of the IO.
- Consideration is given to achieve a balance between new and experienced Members when determining which appointments will be renewed.
- No specific attendance requirements are delineated, however it is required that Committee Members demonstrate a genuine interest and commitment to the purpose of the Committees.
- No method for removal is delineated, as all Members are appointed and serve at the discretion of the IO.
Ex Officio Members
An ex officio member is defined as a member who serves by virtue of an office or position held. An ex officio member may be appointed by the IO as a voting member, a non-voting member, or an alternate member.
- The University Biosafety Officer will serve ex officio as a regular member with full voting privileges.
- The RPO Director will serve as an ex officio member. This individual may serve as an alternate member if so appointed.
- Additional ex officio or alternate members may be appointed at the discretion of the IO.
Alternate Members
An alternate member is defined as a member who substitutes for a specific member or members with similar qualifications, experience or membership category. When an alternate member substitutes at a meeting, they appear as “substitutions” on the minutes. Alternate members may be appointed under the following conditions:
- Must be appointed by the IO and listed in IBC rosters submitted with regulatory documents.
- Must be designated to serve as the alternate for a specific member or members who have the same attributes (e.g. scientific member can only substitute for another scientific member.)
- Alternates must receive same onboarding training as non-alternate members.
- Must receive all proposal materials in advance of the meeting for review if they will be voting during the meeting.
- Alternate members are advised to "vote their conscience" as opposed to representing the position of the regular member for whom they serve.
- If both the regular voting and alternate member both attend a meeting, only the regular voting member may vote. An alternate member may only be required to vote when necessary to achieve or maintain quorum.
Committee Composition
At a minimum the Committee will consist of not less than five regular voting Members and at a minimum will include the following:
- One or more individuals with expertise in recombinant DNA technology, infectious agents, and/or biological safety, and/or physical containment.
- At least two members who are not affiliated with UVM (including family members) and who represent the interests of the surrounding community with respect to health and protection of the environment.
- At least one individual with expertise in plant, plant pathogen, or plant pest containment principles before reviewing and approving experiments involving plants with biosafety level 2 or greater.
- At least one scientist with expertise in animal containment principles before reviewing and approving experiments involving animals with biosafety level 2 or greater.
- A Biological Safety Officer has been appointed. This is a NIH required appointment based on current BSL-3 activities.
In addition, it is recommended that the membership include:
- An individual representing laboratory technical staff;
An individual who meets the requirements of more than one of the categories detailed above may fulfill more than one requirement, if the minimum number is met.
Consultation
The Committees may, at their discretion, obtain consultation from individuals with expertise in specialized areas, such as gene therapy or biosecurity, to assist in the review of complex issues which require expertise beyond or in addition to that available on the IBC. The IBC shall consult with General Counsel and other University Officials, as indicated, to address issues pertaining to institutional policies, applicable law, and standards of conduct and practice. These individuals are not allowed to vote.
General liability insurance coverage
Actions by members carried out as a function of their Committee appointments are included under the University's general liability insurance coverage.
Remuneration
Regular membership on the IBC will be without monetary compensation.
Legal Implications of Membership
Actions by the Members carried out as a function of their Committee appointments are included under the University's general liability insurance coverage.
Conflict of Interest
If an IBC Member has a conflicting interest in a protocol (including, but not limited to being a principal investigator, a co-investigator, or a consultant on that protocol), that Member may only provide information as requested by the IBC and will not be assigned to officially review nor vote on that protocol.
Code of Conduct For Committee Members
This Code of Conduct is a set of behavioral expectations intended to assure that our Committee members uphold the highest level of integrity and ethical standards. Members must not discuss, disclose, or reproduce any protocol-related information, except as necessary to carry out responsibilities or as required by law. Members must limit their electronic access to that which is required to fulfill their Committee duties. Members must never access any research protocols to satisfy personal interest or curiosity. Any printed materials for review should be returned to the IBC or shredded after use.
Committee Member Training
New Members meet with the Chair and the IBC analyst for training and basic introductory materials are provided to acquaint the new Member with the Committee and its functions prior to participation in a meeting. Additional materials are provided for reference during protocol review.
Documentation of Training Completion
Records of completion dates are maintained in RPO shared folder.
Continuing Education
Continuing education is accomplished by retaking the required IBC CITI tutorials at least once every three years, attendance at webinars, regional or national meetings and conferences. Additional education is provided as topics discussed during the monthly Committee meetings.
Revised: 08/13/2024
1.5 Public Records and Open Meetings
Federal Freedom of Information Act (FOIA)
The Freedom of Information Act (FOIA) is a federal law that generally provides that any person has a right, enforceable in court, to obtain access to federal agency records. FOIA applies only to federal agencies. Each state has its own public access laws that are consulted for requests for access to state and local records. Vermont and in turn the University has its own laws and process for records.
Vermont Public Records Act
The University of Vermont is a public body subject to the Vermont Public Records Act (1 V.S.A. §316)(a). Under this law, any person may inspect or copy any public record of a public agency. The definitions of public agency; public records and documents are included in 1 V.S.A. §317.
However, research data and protocols are exempt from disclosure under Vermont law §317(c)(23). Because these records are exempt from public disclosure, the FOIA cannot be used to inspect or copy records. Researchers should forward any request for inspection or copies of research records to the Vice President for Executive Operations according to the UVM Records and Document Request policy.
Revised: 4/12/22
1.6 Electronic Review and Signatures
All reviews, initial, continuing and amendments are reviewed electronically by the IBC members as assigned. Members receive an email notice that a review is pending. They proceed to the electronic record by either utilizing a link embedded within the email or they proceed directly to the system home screen where they will find their review task(s). Members are required to authenticate into Click using their UVM NetID and password prior to completing their review. Each electronic review is stamped with the name of the individual carrying out the review activity (electronic signature), and the time and date that the electronic signature was applied to the review.
Revised 04/19/19
Section II: Research Guidance
2.0 Operations of the IBC
The IBC will follow these operating procedures. These procedures will be reviewed periodically, at least once every three years, to ensure compliance with all pertinent laws governing the use biohazardous materials. Changes to these operating procedures will be developed in consultation with the RPO and the UVM Environmental Health and Safety Office and will be reported to the Institutional Official.
The institution will provide the IBC with resources, office space, professional staff, and support staff sufficient to carry out their responsibilities efficiently and effectively and to serve as day-to-day liaison with appropriate University administrative offices, project investigators, other institutional safety and ethics boards, and various regulatory and funding agencies.
Convened Meetings
The Committees meet monthly if there are agenda items.
Meeting Notices
Meetings are noticed on the IBC website. The agenda, including the time and location of the meeting, are distributed in advance to all members. All pre-meeting protocol materials are located within the electronic submission and review system. All members have access to this system.
Conducting Initial and Continuing Review
Initial and continuing IBC reviews and approvals of a Master Protocol Registration (MPR) will occur in compliance with NIH Guidelines. Continuing reviews will be preceded by IBC receipt of an appropriate progress report from the investigator. Reviewer forms and internal checklists are utilized as a guide to ensure that these criteria have been met.
Review Decisions and Process for Appeal
Review decisions will be forwarded to the researcher in writing. Review decisions may not be overridden. However, the Vice President for Research Administration shall have the final authority to disapprove, restrict or terminate a study which has received IBC approval. There is no process delineated for appeal of Committee decisions. However, there is no prohibition for resubmission of specific requests or protocols for additional review by the Committee.
Protocols Requiring More Frequent Review
Determination of which studies require review more often than annually is done at the time of protocol review, on a case by case basis, depending upon protocol specific factors, including, but not limited to, the level of risk.
Modification to a MPR
The IBC requires that changes in approved research be reviewed prior to initiation. Changes implemented prior to Committee approval is considered noncompliance.
Accidents/Incidents
The IBC is required to report significant problems, violations of the NIH Guidelines, or any significant research-related accident or illness by the Institution. See section 4.4 for further details.
Removal from Consideration
RPO staff will have the authority to remove from further Committee consideration, a study which has obtained initial approval, when the PI fails to respond to ongoing clarifications or training requests. The PI will be required to close the study in UVMClick-IBC.
Documentation
The IBC, through the administrative staff, is responsible for reporting findings, actions as well as requesting clarifications to the investigators in writing, and to the appropriate offices within the institutions’ administration through reports and meeting minutes to institutional officials through their representatives on the Committee, and to sponsors of research, if so required.
Authority to Sign IBC Documents
a. Results of Reviews, Actions and Decisions whether Full or Expedited
Depending upon the nature of the required conditions, the IBC designates any of the following individuals or groups of individuals to determine that the conditions of approval have been satisfied:
- The IBC chair;
- Another member;
- An IBC Analyst/Member or;
- Other qualified IBC administrative staff person, who need not be an IBC member.
Individuals designated by the IBC must have appropriate expertise or qualifications. For some conditions, the review of response materials from investigators will require scientific or other technical expertise. In such cases, the IBC Chair will review the responsive materials or designate another individual who has the appropriate expertise.
Follows are some examples of conditions in which IBC Analysts/Members have been designated to review and approve response materials.
- Correction of minor grammatical and typographical errors in the MPR;
- Requirement to revise the protocol in a specific manner as dictated by the Committee.
b. Administrative Review and Approval
Administrative items are reviewed and approved by IBC Analysts/Members or appropriate IBC staff. IBC Analysts/Members may consult with the Committee chair prior to approval. Below are examples of administrative items.
- Correction of omission of sponsor
- Changes to Key Personnel
- Continuing Reviews (IBC Analyst)
c. Routine Internal Correspondence
Any action, letter, memo or e-mail between the Committee or IBC staff and the faculty or staff of the University that provides information concerning the review of research protocols by the Committee or IBC staff and which do not imply or appear to imply approval of this activity may be signed by the staff member.
d. Decisions Made by the Chair
Any letters, memos or email sent representing the decision or opinions of the Chair or Vice Chair of the IBC, as long as such correspondence does not imply review and approval, may be signed by IBC staff if so designated by the IBC.
Electronic Reviews
All reviews, initial, continuing reviews and modifications are completed electronically by the IBC members as assigned. Members receive an email notice that a review is pending. Members are required to authenticate into the electronic system using their UVM NetID and password prior to completing their review. The system validates the member’s authentication credentials based upon the member’s role in the system and determines available actions for each person. The electronic review is stamped within the system with the name of the individual carrying out the review activity (electronic signature), and the time and date that the electronic signature was applied to the review.
Members only access records that they have been assigned to review.
- Voting Requirements
- Definition of Quorum: A majority of the total number of regular voting members will constitute a quorum. If less than a majority of the total number of regular voting Members is present, if an ex officio nonvoting member(s) is available, that individual may be included to constitute a quorum. If a quorum is lost at any time during the meeting, the meeting shall be adjourned and no further action taken until a quorum is attained.
- Official Committee action (non-exempt) on new MPRs involving biohazardous materials will be by formal vote (exceptions previously noted under E.4) at convened meetings of a quorum of Committee members.
- All meetings will be conducted using Robert’s Rules of Order as guidance, with deviations made as deemed appropriate by the Chair.
- Selection of specific protocols for review by members is determined by the Committee's administrative staff and/or Chair. If an IBC member has a conflicting interest in a MPR (including, but not limited to being a principal investigator or a co-investigator) that member may only provide information as requested by the IBC and will not be assigned to officially review nor vote on that protocol.
- IBC members may participate in a convened meeting of the IBC via telephone or video conferencing. Those members have access to the research protocol materials in advance of the meeting within UVMClick-IBC.
IBC Record Requirements
The IBC keeps all records in accordance with all pertinent regulations. This record keeping includes the following.
Membership rosters
The institution is required to maintain a current list of IBC members identified by name; earned degrees; representative capacity; indications of experience such as board certifications or licenses sufficient to describe each member's chief anticipated contributions to IBC deliberations; and any employment or other relationship between each member and the institution, for example, full-time employee, part-time employee, member of governing panel or board, stockholder, paid or unpaid consultant. UVM rosters indicate regular voting versus alternate members, as well as alternate replacement assignments. Rosters are updated each time there is a change in the Committee membership. Copies of curriculum vitae are obtained and kept on file for all primary and alternate members.
Written procedures and guidelines including, but not limited to, the IBC Policy and Procedure Manual and all website content.
Minutes of meetings document
- date and place of meeting,
- motion to approve prior minutes,
- attendance of members, consultants, and guests,
- a record of members who participated in the convened meeting via phone or video conferencing,
- members/consultants/guests arriving and leaving the meeting once the meeting is called to order,
- which alternate member is replacing a primary member,
- monthly reports from RPO and EHS,
- major points of protocol discussion,
- basis for requiring changes in research or disapproving research,
- motion, including the biosafety containment level, the NIH Guidelines section as applicable, training status, and additional safety requirements as necessary,
- record of the votes (for, against, recused, abstained),
- names of members who recuse themselves secondary to a conflict of interest,
- alternate voting members, and
- time of meeting adjournment.
Approved minutes will be approved by the Committee the following month and signed by the Chair or designee, scanned, and maintained as a PDF in a shared electronic file.
Non-Committee Business Report is available in the UVMClick-IBC system.
Protocol/MPR files are in electronic form in the IBC-Click software. The electronic files include protocols, continuing reviews, amendments, risk assessments, and reported incidents.
Communications to and from the IBC are maintained in the electronic protocol file.
Minutes under NOT-OD-25-082 Promoting Maximal Transparency Under the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules, will be posted to our IBC webpage. All recorded meeting discussion will be included with the exception of protocols that do not have federal sponsorship and/or biotechnical companies under a NDA with the University.
Revised 07/21/2025
2.1 Types of Committee Review
The IBC will review and have authority to approve, require modifications in (to secure approval), or disapprove all research and teaching activities involving biohazardous materials. These review categories are employed for new master protocol registrations (MPR), amendments to add new projects to existing registrations, and continuing review.
Preliminary Reviews
In all cases, there are two preliminary reviews that require completion prior to review by the Committee.
Biological Safety Team Review
New master registrations and amendments with new projects will be forwarded to the biosafety team for recommendations on risk mitigation practices relevant to the proposed research or teaching activity. The biosafety team’s risk assessment document and feedback will be appended to the electronic MPR.
In the event there is insufficient time for a biosafety team member to conduct a pre-review within the posted submission deadlines, the protocol will be placed on an agenda and the IBC will conduct the review after completion of an IBC Analyst Pre-Review. The biosafety team member is also an IBC member and will be part of the discussion. A post-approval monitoring visit to assess operating procedures may be stipulated depending upon the outcome of the IBC’s review.
IBC Analyst Pre-Review
The IBC must receive sufficient information regarding proposed activities to make the determinations as required by regulations. Before placing a submission on the agenda, the IBC Analyst will conduct a pre-review of submitted materials. A specific regulatory checklist is followed to determine if the submission is ready for review by the IBC. If the submission is incomplete or lacks information necessary to conduct a review, it will not be reviewed until the information is provided. Once it is determined that the submission is satisfactory, the review will continue.
Committee Review (convened meeting)
The IBC employs the convened meeting review process for review and approval of submissions that propose to use non-exempt biohazardous materials in a risk group category greater than or equal to RG-2 and/or requiring Biosafety Level 2 (BSL2) containment practices. Research or teaching activities involving RG-1 agents and/or recombinant or synthetic nucleic acid molecules may also require review by the IBC depending on the nature of the proposed work.
Notification to Research Community
Committee meetings are noticed on the Committee website. Submissions are added to an Agenda on a rolling basis after preliminary pre-reviews are complete.
Notification to the Committee
Distribution of the Agenda to all members occurs approximately 2 weeks in advance of the convened meeting. All new MPRs, amendments, incidents and other business requiring full committee action are placed on the Agenda for discussion.
Access to the MPR Materials
Committee members have access to all of the MPR materials through the electronic submission and review system.
Reviewer Assignments
Primary and Secondary Reviewers are assigned by the RPO Analyst to review the submission and all provided materials. Efforts are made to match the primary reviewer’s expertise to the project’s subject matter. Reviewers are encouraged to contact the PI to resolve/clarify major concerns prior to the meeting . The reviewers summarize the proposed research or teaching activities for the full Committee at a convened meeting and answer questions during the discussion. An approval memo or a request for further clarification will be sent to the principal investigator.
Designated Member Review
The regulations have flexibility to allow for a designated member review process. A new MPR or amendment to add a new project to an existing MPR may be reviewed by a designated member of the IBC under one or more of the following conditions:
- The proposed work is equivalent to work already approved under the existing registration, as determined by the IBC, and there are no substantive differences that would change the biosafety and or public health considerations for the proposed project.
- The proposed work receives a preliminary category of BSL1.
Assigning Reviewers
The submission will be assigned to a designated member for review once the preliminary reviews are complete. The Chair reserves the right to also provide the materials to each member of the Committee for review. If no additional clarifications or requests for a Full Committee review at a convened meeting are received, an approval will be signed at the discretion of the Chair. An approval memo or a request for further clarification will be sent to the principal investigator.
Review Simultaneous with Initiation
The regulations allow for review simultaneous with initiation of the project if the experiments are designated as BSL1 and fall under Section III-E of the NIH Guidelines. In these instances, researchers will follow the designated review process listed above.
Exemption Determinations
The regulations allow exemption from IBC review for experiments falling within Section III-F. Though these projects may be exempt from the Guidelines, they still need to be forwarded electronically to the IBC for a formal determination and review of other Federal, State, and local biosafety standards that may apply. The Chair will make the determination of exemption.
Section III-F. Exempt Experiments
The following recombinant or synthetic nucleic acid molecules are exempt from the NIH Guidelines and registration with the Institutional Biosafety Committee is not required; however, other federal and state standards of biosafety may still apply to such research (for example, the Centers for Disease Control and Prevention (CDC)/NIH publication Biosafety in Microbiological and Biomedical Laboratories).
Section III-F-1. Those synthetic nucleic acids that: (1) can neither replicate nor generate nucleic acids that can replicate in any living cell (e.g., oligonucleotides or other synthetic nucleic acids that do not contain an origin of replication or contain elements known to interact with either DNA or RNA polymerase), and (2) are not designed to integrate into DNA, and (3) do not produce a toxin that is lethal for vertebrates at an LD50 of less than 100 nanograms per kilogram body weight. If a synthetic nucleic acid is deliberately transferred into one or more human research participants and meets the criteria of Section III-C, it is not exempt under this Section.
Section III-F-2. Those that are not in organisms, cells, or viruses and that have not been modified or manipulated (e.g., encapsulated into synthetic or natural vehicles) to render them capable of penetrating cellular membranes.
Section III-F-3. Those that consist solely of the exact recombinant or synthetic nucleic acid sequence from a single source that exists contemporaneously in nature.
Section III-F-4. Those that consist entirely of nucleic acids from a prokaryotic host, including its indigenous plasmids or viruses when propagated only in that host (or a closely related strain of the same species), or when transferred to another host by well-established physiological means.
Section III-F-5. Those that consist entirely of nucleic acids from a eukaryotic host including its chloroplasts, mitochondria, or plasmids (but excluding viruses) when propagated only in that host (or a closely related strain of the same species).
Section III-F-6. Those that consist entirely of DNA segments from different species that exchange DNA by known physiological processes, though one or more of the segments may be a synthetic equivalent. A list of such exchangers will be prepared and periodically revised by the NIH Director with advice of the RAC after appropriate notice and opportunity for public comment (see Section IV-C-1-b-(1)-(c), Major Actions). See Appendices A-I through A-VI, Exemptions under Section III-F-6--Sublists of Natural Exchangers, for a list of natural exchangers that are exempt from the NIH Guidelines.
Section III-F-7. Those genomic DNA molecules that have acquired a transposable element, provided the transposable element does not contain any recombinant and/or synthetic DNA.
Section III-F-8. Those that do not present a significant risk to health or the environment (see Section IV-C-1-b-(1)-(c), Major Actions), as determined by the NIH Director, with the advice of the RAC, and following appropriate notice and opportunity for public comment. See Appendix C, Exemptions under Section III-F-8 for other classes of experiments which are exempt from the NIH Guidelines.
Administrative Review/Approval
IBC staff will review and approve study team member changes and continuing reviews.
Revised 08/13/24
2.2 IBC Review Determinations
The IBC shall review and have authority to approve, require modifications in (to secure approval), or disapprove all research and teaching activities covered by this policy, including exempt research activities.
APPROVED
This decision indicates that the initial review of a master protocol registration (MPR) at a convened meeting or via designated review has been approved without requiring either further (a) changes to the MPR or (b) submission of clarifications or additional documents.
CLARIFICATIONS REQUESTED FOR INITIAL APPROVAL
This decision indicates approval is pending satisfactory resolution of conditions or clarifications that the IBC requires to approve the MPR. Under this scenario, for full review MPRs, further review by the IBC at a subsequent convened meeting is not necessary to secure final approval.
The designated review process is employed to review the response from the investigator. The IBC directs that the IBC chairperson (or other individual(s)) to review and determine on behalf of the IBC whether the changes, clarifications, and/or additional documents to be submitted by the investigator(s) are satisfactory.
If under the designated review of the response, the IBC chairperson (designated reviewer) is unable to approve the MPR because he/she cannot make the determinations required for approval, they can either refer the project to the IBC for further review and action at a convened meeting, or defer approval of the MPR and require that the investigator (a) make changes to the MPR, or (b) submit clarifications or additional documents prior to further review by the IBC chairperson (or designated reviewer(s)).
DEFERRED (tabled)
This action is taken under full Committee review any time the IBC cannot make one or more of the determinations required for approval but with major revisions may be found to be approvable. When deferring a project, the IBC, under its authority to require modifications in order for an investigator to secure approval, may require that the investigator (a) make changes to the MPR or (b) submit clarifications or additional documents. The research may not proceed until the IBC reviews, at a subsequent convened meeting, the revised MPR and approves it.
DISAPPROVED
This action is taken under full Committee review when the determinations required for approval of the research cannot be made, even with substantive clarifications or modifications to the MPR. If the IBC decides to withhold approval of a MPR, it will include in its written notification a statement of the reasons for its decision.
The above review determinations also apply to follow on submissions to an approved MPR including amendments and continuing reviews.
ADMINISTRATIVE HOLDS, SUSPENSIONS OR TERMINATIONS
All currently approved research is subject to modification or change in approval status, as deemed necessary by the IBC. The IBC has the authority to suspend or terminate research for not being conducted in accordance with IBC requirements or federal regulations; or if it has been associated with unexpected harm to personnel or the environment. The Investigator also has the option to place the research on administrative hold.
Administrative Hold
An administrative hold is a voluntary action by an investigator to temporarily or permanently stop some or all approved research activities. Administrative holds are not considered suspensions or terminations, and do not meet reporting requirements to the Office of Science Policy. Administrative holds must not be used to avoid reporting deficiencies or circumstances that otherwise require reporting to federal agencies. The Principal Investigator may place an administrative hold on research if; a complaint is received; an allegation of non-compliance is reported to the IBC; or a discovery by the investigator of new potential risk to personnel or the environment. The Investigator must notify the IBC of the hold through an amendment.
During administrative hold, the research remains subject to continuing review and requirements for reporting non-compliance and incidents.
The IBC designee may make recommendations for additional education and/or compliance interventions for the Investigator and research personnel as the result of an administrative hold. At any point, the IBC Committee can suspend the research, which will result in required regulatory reporting.
Suspension
A suspension of IBC approval is a directive of the convened IBC, Biosafety Officer or IBC designee either to stop temporarily some or all previously approved research activities, or to stop permanently some or all previously approved research activities. All suspensions are immediately reportable to federal agencies.
The Biosafety Officer has the authority to suspend previously approved research when required for the urgent protection of personnel and/or the environment and insufficient time exists for the convened IBC to review the issue. Any suspension of research by the above individuals is placed on the next available agenda of a meeting of the full IBC. The convened IBC will review the circumstances that led to the suspension and will make the determination to uphold or overturn the suspension. The IBC will recommend corrective actions if the suspension is upheld.
During suspension, the research remains subject to continuing review and requirements for reporting non-compliance and incidents.
The IBC Chair, Biosafety Officer, or designee notifies the investigator of the suspension and the reason for the suspension.
Termination
A termination of IBC approval is a directive of the convened IBC or IBC designee to permanently stop all previously approved research activities. All terminations are reportable to federal agencies. MPR approval will not be terminated without first undergoing temporary suspension and completion of a review following the Noncompliance Policy and Procedures process.
Previously approved research may only be terminated by the convened IBC, including MPRs originally approved under designated procedures.
Terminated protocols are considered closed and no longer require continuing review.
Revised 05/25/23
2.3 Coordination with Other Compliance Committees/Divisions/Cores
Animal Use (IACUC)
Review by the IBC is independent of the review by the Institutional Animal Care and Use Committee (IACUC). However, there is representation on both committees that communicate and coordinate regarding Biosafety issues in animal studies. Initiation of the animal component of the study is contingent upon the completion of and approval of IACUC process.
Human Subjects (IRB)
Review by the IBC is independent of the review by the Institutional Review Board (IRB).
Sponsored Project Administration (SPA)
SPA requires documentation of protocol approvals prior to release of funds. IBC staff share protocol statuses with SPA for this purpose.
Institutional Review Entity (IRE) Subcommittee
The IRE is a subcommittee of the IBC. Researchers who believe that they may be conducting DURC are required to submit a MPR to the IBC for review and initial determination. The IBC and IRE work closely to ensure proper oversight of DURC. See Section 3.5 for the organizational framework for oversight of DURC.
UVM Core Facilities
An ancillary review will be sent to the director of each core facility when an MPR indicates their use.
Revised 05/25/23
2.4 Conducting IBC Business in the Event of a Pandemic or other Significant Emergency
IBC Meetings
Internal policies state that convened meetings are required for 1) initial review of Master Protocol Registrations (MPR) that propose the use of non-exempt biohazardous materials in a risk category greater than or equal to Biosafety Level 2 (BSL2); 2) any amendments that have the potential to increase Biosafety Level; 3) determinations of level of seriousness for noncompliance cases. While optimal, there is no requirement to conduct the convened meeting in person. Use of teleconferencing or audio/video conferencing is permissible. If quorum cannot be achieved, convened meetings will be postponed until enough members can be present.
Security of IBC remote meetings will be assured by using only University-approved videoconferencing software logging in only with UVM credentials. Guest presence will be controlled by the meeting owner which, is typically a RPO staff person. IBC videoconference meetings will not be recorded. Quorum of members will assured by a count of those in attendance prior to opening the meeting and a second time after moving into open session prior to the protocol vote.
Minutes of meetings will be captured following current methods for in-person meetings. The manner of engagement of each member will be noted (e.g. in-person, telephone, video.) Votes to go in or out of sessions, as well as to vote for specific registrations, will occur by the Chair asking for members who approve an order of business by asking “All approved say aye”, “All opposed say nay”, “All abstaining say aye”. Members participating through video conferencing can also use the chat feature to add comments to the discussion. Members with conflicts will sign out of the meeting during the vote and IBC staff will invite them back into the meeting when the conflicting MPR discussion is complete. Meeting guests will be invited during discussion of their registration and signed out once that discussion is completed.
MPR Review
New BSL2 MPRs or amendments to MPRs representing a potential for an increase in biosafety level must be reviewed in a convened meeting. Convened meetings can proceed as described above.
New BSL1 MPRs or amendments to existing MPRs which do not change the safety level can continue to be reviewed through the current designated review process.
MPR review documentation will be through the UVMClick-IRB electronic MPR submission software.
Biosafety Team Risk Reviews
The biosafety team member’s review of risk will continue to be conducted when new work is proposed as long as the laboratory remains in operation.
Post Approval Monitoring
Post Approval Monitoring visits will be placed on hold until normal working conditions are in place.
Training
If personnel refresher training expires during the emergency, personnel are allowed to continue their work as long as they have received previous training and have demonstrated proficiency. There will be no consequences; however, once working conditions have been restored, personnel must complete refresher training in accordance with the IBC policy.
Pause in Research Activities
If laboratory activities are required to be placed on pause secondary to institution-wide policy to address a public health situation, the IBC does not require notification in the pause of work.
Revised: 07/29/2024
3.0 Master Protocol Registration Submission
Researchers may now submit one master protocol registration (MPR) to cover multiple projects with similar biosafety levels. Additionally, teaching laboratories that utilize biohazardous materials must submit a MPR describing the work and safe handling procedures being conducted in the teaching laboratory. Adopting this model will reduce regulatory burden. Researchers/professors will combine multiple teaching, or internally- and externally-funded projects under one MPR. Laboratory activities requiring BSL-1 and BSL-2 work practices may be combined.
A/BSL-3 or BSL-3 research and research meeting criteria for Dual Use Research of Concern (DURC) or Pathogens with Pandemic Potential (PPP) require individual master protocol registrations due to their complexity and different regulatory requirement from BSL-1 and BSL-2.
Since the MPRs are grouped together based upon biosafety levels, it is important that this level is established early in the review process for a new project. The following tools can help researchers make an initial determination of the appropriate risk group, containment level and work practices.
The NIH/CDC BMBL 6th edition will help get you started in making a risk assessment.
The American Biological Society Association maintains an excellent reference for risk groups.
Implementation Guidance for the USG for Oversight of Dual Use Research of Concern and Pathogens with Enhanced Pandemic Potential (DURC/PPP). You may use the DURC/PPP self-determination tool to Assess whether your research meets the criteria for Category 1 or Category 2 research under the new USG DURC/PPP policy.
Revised 03/18/2025
3.1 Initial PI Risk Assessment
It is the responsibility of all PIs to provide an initial assessment of the risk factors and risk levels involved in their proposed activities. In many instances, the PI has significant experience working with similar biological agents or recombinant DNA and is in the best position to estimate the appropriate biosafety level (BSL-1, BSL-2 or BSL-3) for the laboratory.
Guidance
The following guidance on risk assessment is provided to assist PI’s in conducting an accurate and effective risk assessment. The steps listed below are modified excerpts from the Biosafety in Microbiological and Biomedical Laboratories (BMBL) 6th Edition publication:
Step 1: Identify agent hazards and perform an initial assessment of risk. Consider the principle hazardous characteristics of the agent, which include its capability to infect and cause disease in a susceptible human host, severity of disease, and the availability of preventive measures and effective treatments.
Step 2: Identify laboratory procedure hazards. The principle laboratory procedure hazards are agent concentration, suspension volume, equipment and procedures that generate small particle aerosols and larger airborne particles (droplets), and use of sharps. Procedures involving animals can present a number of hazards such as bites and scratches, exposure to zoonotic agents, and the handling of experimentally generated infectious aerosols.
Step 3: Make an [initial] determination of the appropriate biosafety level and select additional precautions indicated by the risk assessment. Note: The IBC will make the final biosafety level determination.
Step 4: Evaluate the proficiencies of staff regarding safe practices and the integrity of safety equipment.
In conducting a risk assessment, the PI should ensure that laboratory workers have acquired the technical proficiency in the use of microbiological practices and safety equipment required for the safe handling of the agent, and have developed good habits that sustain excellence in the performance of those practices.
Revised 08/13/24
3.2 Risk Assessment
The ongoing practice of biological risk assessment is the foundation of safe laboratory operations. Risk assessment requires careful judgement and is an important responsibility for principal investigators (PIs). Institutional leadership and oversight resources, such as Institutional Biosafety Committees (IBC), animal care and use committees, biological safety professionals, occupational health staff, and laboratory animal veterinarians also share this responsibility.
The institution supports researchers by requesting that a biosafety team member review new protocol submissions (e.g. initial master protocol registrations and amendments) to assess if the risk group and biosafety level are accurate and to make suggestions to improve procedures for handling biohazardous materials.
If there is insufficient time for a biosafety team member to conduct a pre-review within the posted submission deadlines, the protocol will be placed on an agenda and the IBC will conduct the review. The biosafety team member is also an IBC member and will be part of the discussion. A post-approval monitoring visit to assess approved operating procedures may be stipulated depending upon the outcome of the IBC’s review.
Note: The UVM IBC reviews applicable protocols involving human subjects who are being treated at The University of Vermont Medical Center (UVMMC). The biosafety team works with specific UVMMC service lines (see gene therapy) to assist with same.
Risk Groups
Agents are classified into four Risk Groups according to their relative pathogenicity for healthy adult humans as follows:
- Risk Group 1 (RG-1) agents are not associated with disease in healthy adult humans.
- Risk Group 2 (RG-2) agents are associated with human disease which is rarely serious and for which preventive or therapeutic interventions are often available.
- Risk Group 3 (RG-3) agents are associated with serious or lethal human disease for which preventive or therapeutic interventions may be available.
- Risk Group 4 (RG-4) agents are likely to cause serious or lethal human disease for which preventive or therapeutic interventions are not usually available.
Risk Factors
The following factors should be considered when conducting a risk assessment and determining the level of containment:
- Pathogenicity of the biohazardous material(s) - Consideration should include disease incidence and severity.
- Route of transmission (e.g., parenteral, airborne, by ingestion) - When planning to work with a relatively uncharacterized agent with an uncertain mode of transmission, the potential for aerosol transmission should be strongly considered.
- Agent stability - Should include a consideration of factors such as desiccation, exposure to sunlight or ultraviolet light, or exposure to chemical disinfectants.
- Infectious dose of the agent and communicability - Consideration should include the range from the healthiest immunized worker to the worker with lesser resistance.
- Concentration - Include consideration of the milieu containing the organism (e.g., solid tissue, viscous blood or sputum, liquid medium) and the activity planned.
- Volume - >10 liters is considered large scale and is subject to further review and higher containment level.
- Origin of the biohazardous material(s) - Consideration should include factors such as geographic location, host, and nature of the source.
- Availability of data from animal studies - This information may be useful in the risk assessment process in the absence of human data.
- Established availability of immunization/vaccine or treatment - The unavailability of immunization/vaccine or treatment may impact the risk involved in the use of biohazardous material(s).
- Gene product effects, such as toxicity, physiological activity, and allergenicity.
Biosafety Level (Biological and Physical Containment Level)
The final risk assessment determination (RG-1 to RG-4) is used to set the appropriate biosafety level (BSL-1 to BSL-4) for the biohazardous material(s). The biosafety level describes the degree of physical containment and biosafety practices required to confine these materials and to reduce the potential for exposure of laboratory workers, persons outside the laboratory, and the environment.
The following is a general description of the biosafety levels as described in the Biosafety in Microbiological and Biomedical Laboratories (BMBL) 6th edition:
- Biosafety Level 1 (BSL-1) is suitable for work involving well-characterized agents not known to consistently cause disease in immunocompetent adult humans, and present minimal potential hazard to laboratory personnel and the environment. BSL-1 laboratories are not necessarily separated from the general traffic patterns in the building. Work is typically conducted on open bench tops using standard microbiological practices. Special containment equipment or facility design is not required, but may be used as determined by appropriate risk assessment. Laboratory personnel must have specific training in the procedures conducted in the laboratory and must be supervised by a scientist with training in microbiology or a related science.
- Biosafety Level 2 (BSL-2) builds upon BSL-1. BSL-2 is suitable for work involving agents that pose moderate hazards to personnel and the environment. It differs from BSL-1 in that 1) laboratory personnel have specific training in handling pathogenic agents and are supervised by scientists competent in handling infectious agents and associated procedures; 2) access to the laboratory is restricted when work is being conducted; and 3) all procedures in which infectious aerosols or splashes may be created are conducted in a biosafety cabinet (BSC) or other physical containment equipment.
- Biosafety Level 3 (BSL-3) is applicable to clinical, diagnostic, teaching, research, or production facilities where work is performed with indigenous or exotic agents that may cause serious or potentially lethal disease through inhalation route exposure. Laboratory personnel must receive specific training in handling pathogenic and potentially lethal agents, and must be supervised by scientists competent in handling infectious agents and associated procedures. All procedures involving the manipulation of infectious materials must be conducted within BSCs, other physical containment devices, or by personnel wearing appropriate personal protective equipment. A BSL-3 laboratory has special engineering and design features.
NOTE: There are also biosafety levels for work with infectious agents in vertebrate animals. For a complete description of the animal biosafety levels, consult the BMBL. https://www.cdc.gov/labs/pdf/SF__19_308133-A_BMBL6_00-BOOK-WEB-final-3.pdf
The biosafety level may be equivalent to the Risk Group classification of the agent or it may be raised or lowered based on the results of the risk assessment. If you have any questions regarding the risk assessment or appropriate containment level, you may consult with the IBC. The IBC makes the final determination of the appropriate biosafety level.
Revised 08/13/24
3.3 Research Including Recombinant DNA or Synthetic Nucleic Acid Molecules
In the context of the NIH Guidelines, recombinant and synthetic nucleic acids are defined as:
(i) molecules that a) are constructed by joining nucleic acid molecules and b) that can replicate in a living cell, i.e., recombinant nucleic acids;
(ii) nucleic acid molecules that are chemically or by other means synthesized or amplified, including those that are chemically or otherwise modified but can base pair with naturally occurring nucleic acid molecules, i.e., synthetic nucleic acids, or
(iii) molecules that result from the replication of those described in (i) or (ii) above.
As a condition for NIH funding of recombinant or synthetic nucleic acid molecule research, institutions shall ensure that such research conducted at or sponsored by the institution, irrespective of the source of funding, shall comply with the NIH Guidelines.
Section III of the NIH Guidelines details the type of experiments covered by the Guidelines and the level of reviewed required.
Revised: 07/24/2024
3.4 Research Including Animals
MPRs which involve the exposure of animals to recombinant and/or synthetic nucleic acid molecules, infectious agents or biotoxins require review by the IBC and the Institutional Animal Care and Use Committee (IACUC) committees. This includes the generation of new strains of genetically-modified animals using transgenic, homologous recombination or genome editing approaches.
The office allows simultaneous reviews with the IBC and IACUC committees. However, to protect animals, the IACUC approval will not be released until IBC has approved appropriate containment conditions required for the animal experiments and animal housing.
Both the IBC and the IACUC will review incident reports and changes in projects submitted during the course of the research.
Revised 03/15/19
3.5 Research Including Infectious Agents
Infectious agents include any biological agents and biologically derived materials that present a risk or potential risk to the health of humans or animals, either directly through infection or indirectly through damage to the environment.
Categories of Potentially Infectious Materials
- Human, animal, and plant pathogens (bacteria, parasites, fungi, viruses, prions).
- All human blood, blood products, tissues, and certain body fluids when used in conjunction with infectious agents or recombinant or synthetic nucleic acid molecules.
- Cultured cells and potentially infectious agents these cells may contain.
- Clinical specimens.
- Infected animal and animal tissues.
Centers for Disease Control and Prevention (CDC)
The CDC has set forth an advisory document, the Biosafety in Microbiological and Biomedical Laboratories (BMBL), regarding best practices for the safe conduct of work in biomedical and clinical laboratories from a biosafety perspective. The IBC uses this document to conduct review of these types of projects. To address the requirements of the BMBL, the principal investigator must:
• Limit or restrict access to the laboratory when work with infectious agents is in progress. The PI must include a determination of who may be at increased risk and appropriately limit or deny access.
• Establish policies and procedures to limit access to those individuals who have been advised of the potential hazards and meet specific entry requirements (e.g., immunization).
• Ensure that laboratory personnel are offered, at no cost, appropriate immunizations or tests for the infectious agents handled or potentially present in the laboratory (e.g., hepatitis B vaccine, tuberculosis skin testing).
• Select and provide appropriate personal protective equipment required for work with biohazardous materials.
• Ensure that laboratory and support personnel receive appropriate training on the potential hazards associated with the work involved, the necessary precautions to prevent exposures, the exposure evaluation procedures, and that personnel receive annual updates or additional training as necessary for procedural or policy changes.
• Develop standard operating procedures incorporating biosafety procedures or a biosafety manual prepared specifically for the laboratory, advise personnel of special hazards, and require them to read and follow instructions on practices and procedures.
Local Requirement for Standard Operating Procedures
For projects including infectious agents and certain viral vectors, a standard operating procedure or an applicable Biohazardous Agent Reference Document (BARD) is required as part of the MPR submission. BARDs were developed by the IBC to assist researchers in development of these procedures. Researcher may use the BARDs as written or the BARDs may be edited as applicable. The standard operating procedure template and BARDs may be found here.
Revised: 07/24/2024
3.5.1 Research Including Adeno-Associated Virus
Adeno-associated virus (AAV) and recombinant adeno-associated virus (rAAV) are commonly used for gene expression with fewer associated biosafety concerns when compared to viral vectors that are persistent and able to integrate into the genome. The IBC will apply the following criteria for determining the appropriate biosafety containment and handling of AAV:
BSL-1 Transgene does not express an oncogene or toxin, viruses generated without helper virus, acceptable verification that helper virus is not present, or propagation in insect cell lines
BSL-2 Transgene that expresses an oncogene or toxin, viruses that are propagated in human cell lines without further purification before use, known presence of helper virus, or lack of acceptable verification of purification
Registrations including rAAV must attach the Adeno-Associated Viral Vector (AAV) BARD with completed Summary of Biosafety Level Requirements for AAV Use (located on the last page of the BARD document).
Revised 05/07/20
3.5.2 Policy on Research Involving Human Materials and Cell Lines
Background
At the present time, the UVM IBC only reviews work involving human-derived materials when used in conjunction with recombinant and/or synthetic nucleic acid molecules, infectious agents, or when used for studies involving animals. The default position of the IBC and Risk Management & Safety has always been that human-derived materials, including primary and established human cell lines, are considered potentially infectious. Because of the potential for contamination with blood-borne pathogens (BBPs), human materials must always be handled using containment practices required for RG-2 agents, thus requiring the use of BSL-2 containment practices for in vitro laboratory work involving human materials/cell lines or ABSL-2 containment practices when human materials/cell lines are used in animals.
BSL-2 and/or ABSL-2 containment practices must be used for the duration of the experiment or until such time steps are taken to inactivate pathogens that may be present in the human materials (e.g., by cell/tissue fixation, extraction, etc.).
IBC Approval of Changes in Containment Practices when Human Materials and Cell Lines are in Use
Under certain circumstances, the PI may request a reduction in biosafety level for work involving certain human materials/cell lines as part of an approved IBC registration. The following conditions must be met for the IBC to consider a reduction from BSL-2/ABSL-2 to BSL-1/ABSL-1:
- The PI must provide documentation demonstrating that the human materials/cell lines are certified to be free of (test negative for the presence of) BBPs.
- The PI must articulate the scientific and biosafety rationale for a reduction in biosafety level and the specific research activities covered by the request.
- This would typically involve research with established human cell lines that are 1) certified as pathogen-free, and 2) obtained from a source providing documentation indicating that the cells may be handled safely using BSL-1 containment practices.
- A risk assessment must be provided by the PI describing the potential health consequences to research personnel of an accidental exposure to the human materials/cell lines. The consequences must be consistent with the proposed use of A/BSL-1 containment practices.
- The use of established human cell lines or other human derived materials in animals (e.g., xenotransplantation studies) requires both IBC and IACUC review. The justification for a reduction to ABSL-1 containment must include an assessment of the impact of zoonotic disease transmission and the susceptibility of animal subjects to infection by human pathogens.
- The PI must articulate the scientific and biosafety rationale for a reduction in biosafety level and the specific research activities covered by the request.
The UVM biosafety team will review the requested reduction and provide recommendation to the IBC for consideration.
Roles and Responsibilities:
The Principal Investigator (PI) is responsible for:
- Obtain IACUC and IBC approval for animal research involving human cell lines and human derived materials
- Ensure that all staff and students, including the PI, who handle human cell lines or human derived materials complete the bloodborne pathogen training
- Provide access to the Hepatitis B immunization program for employees
- Notifying OACM of implantation or injection of human cell lines/tissues into research animals by labeling cages with biohazard treatment cards with the date and time of injection
Environmental Health and Safety/Biosafety team is responsible for:
- Oversight of the University’s Bloodborne Pathogen Program, including annual training for those conducting research with human-derived materials.
- Recommendations regarding PI risk assessment
The IACUC is responsible for:
- Approve research with human cell lines or human derived materials injected or implanted into laboratory animals.
The IBC is responsible for:
- Approve research with human cell lines or human derived materials when used in conjunction with recombinant and/or synthetic nucleic acid molecules, infectious agents, or injected or implanted into laboratory animals
- Establish biocontainment levels and special handling requirements based upon the risk assessment conducted by the BSO.
Laboratory Animal Resources (OACM) provides: https://oacm.w3.uvm.edu/
- Provide hands-on animal handling training in the Animal Care Facilities
- Provide PPE for use by researchers
- Provide labels for use by researchers to flag animals exposed to human‐derived materials
- Provide ABSL-2 animal room access (once hands-on training has been completed)
Revised: 07/24/2024
3.6 Research Including Biotoxins
Biological toxins can include metabolites of living organisms, degradation products of dead organisms, and materials rendered toxic by the metabolic activity of microorganisms. Some toxins can also be produced by bacterial or fungal fermentation, by the use of recombinant and synthetic nucleic acid molecule technology, or by chemical syntheses of low molecular weight toxins. Protocols utilizing biotoxins must be reviewed by the IBC prior to use.
Visit the Center for Disease Control and Prevention for a list of biotoxins.
Revised 07/30/25
3.7 Select Agent Program at UVM
In Winter of 2025, the sole select agent (brucella) being studied at UVM was removed from the Select Agent Toxin list, therefore UVM deregistered with the Federal Select Agent Program.
Revised 03/28/25
3.8 Research Including Human Subjects
IBC will interact with the Institutional Review Board (IRB) in the review of research that includes human research participants; including, but not limited to, Recombinant DNA/Human Gene Transfer (HGT) and research with biologically derived toxins or infectious agents. In general, the IBC advises the IRB on risk assessment and biosafety issues according to the NIH guidelines.
Process to Obtain Approval
Food and Drug Administration Approval
FDA approval is required prior to any institutional review. Feasibility approvals are required from the following hospital services, regardless of sponsorship.
Investigational Drug Pharmacy
Environmental Health and Safety Department
Infectious Disease
Infection Prevention
Each of these services lines will need to see the following materials:
Clinical Trial Protocol
Investigational Drug Brochure
Standard Operating Procedures for Drug Preparation
Standard Operating Procedures for Transport of the Drug
Standard Operating Procedures for Handling Drug by Clinical Staff
IBC Review
Once all service lines have agreed that the project can be conducted, the MPR can be submitted to the IBC for review. Data entry of the project into Click to include the following uploads:
Hospital service line approvals;
Clinical Trial Protocol;
Investigational Drug Brochure;
Informed consent draft.
IRB Review
Once the IBC has provided a provisionary approval, the human subject protocol can be submitted for review by the Institutional Review Board.
Note: All previous requirements to obtain approval from the Recombinant DNA Advisory Committee and to register with NIH no longer exist as of August 2018.
Revised 03/07/19
3.9 Projects Involving Plants
Projects involving plants require review by the IBC committee. Consultants may be called upon to address these types of protocols.
Revised 03/15/19
3.10 Dual Use Research of Concern & Pathogens with Enhanced Pandemic Potential
Despite its value and benefits, certain types of research conducted for legitimate purposes can be utilized for both benevolent and harmful purposes. Such research is called “dual use research.” Dual use research of concern (DURC) is life sciences research that, based on current understanding, can be reasonably anticipated to provide knowledge, information, products, or technologies that could be directly misapplied to pose a significant threat to public health and safety, agricultural crops and other plants, animals, the environment, material or national security.
The “U.S. Government (USG) Policy for Oversight of Life Sciences Dual Use Research of Concern” articulates the practices and procedures required to ensure that dual use research of concern is identified at the institutional level and risk mitigation measures are implemented as necessary.
Designation of an Institutional Contact for DURC (ICDUR)
The Executive Director for Research Administration is designated as the institution’s ICDUR. The ICDUR serves as the internal resource for issues regarding compliance with and implementation of the requirements for the oversight of research that falls within the scope and/or meets the definition of DURC. If questions arise regarding compliance, implementation of this Policy or when guidance is needed about identifying DURC or developing risk mitigation plans, the ICDUR serves as the liaison (as necessary) between UVM and the relevant program officers at the Federal funding agencies, or for non-Federally funded research, between UVM and NIH (or the appropriate Federal funding agency to which NIH refers the institution).
Initial Identification and Review of Dual Use Research of Concern
The USG has limited the scope to a well-defined subset of life sciences research that involves 15 agents and toxins and seven categories of experiments.
Agents and Toxins
| Avian influenza virus (highly pathogenic) | Marburg virus |
| Bacillus anthracis | Reconstructed 1918 Influenza virus |
| Botulinum neurotoxin | Rinderpest virus |
| Burkholderia mallei | Toxin-producing strains of Clostridium botulinum |
| Burkholderia pseudomallei | Variola major virus |
| Ebola virus | Variola minor virus |
| Foot-and-mouth disease virus | Yersinia pestis |
| Francisella tularensis |
Categories of Experimental Effects
- Enhances the harmful consequences of the agent or toxin
- Disrupts immunity or the effectiveness of an immunization against the agent or toxin without clinical and/or agricultural justification
- Confers to the agent or toxin resistance to clinically and/or agriculturally useful prophylactic or therapeutic interventions against that agent or toxin or facilitates their ability to evade detection methodologies
- Increases the stability, transmissibility, or the ability to disseminate the agent or toxin
- Alters the host range or tropism of the agent or toxin
- Enhances the susceptibility of a host population to the agent or toxin
- Generates or reconstitutes an eradicated or extinct agent or toxin listed above.
How Dual Use Research of Concern is Confirmed
All projects involving biohazardous materials are submitted through the electronic submission system. The electronic form includes specific questions to help assess whether a project may be dual use research. When research is identified by a PI as utilizing one of the listed agents or toxins, the project will be initially reviewed by the IBC Chair and the Biosafety Officer who will make the following determinations:
- Verification that research utilizes one or more of the agents or toxins listed;
- Determination of whether the research produces, aims to produce, or is reasonably anticipated to produce one or more of the effects listed;
- Assessment of the dual use risks and the benefits of the research;
- Review proposed risk mitigation plan for DURC, as necessary.
If determined by the initial review that the project is not DURC, then the project will continue through the regular review process. If it is determined to be DURC, the project will be referred to the Institutional Review Entity (IRE). See flow chart below.
Institutional Review Entity (IRE) Review
The full IRE consists of the UVM Biosafety Officer, the Chair of the IBC, the Director of The Research Protections Office, the Assistant Director for Health & Safety, the Executive Director for Research Administration, a community member, representatives of the scientific community, and an individual from the State Department of Health. When appropriate, representatives from Police Services, University Communications, or a representative of the Office of the General Counsel will be consulted as needed. The IRE functions as follows:
- With sufficient empowerment by the institution to ensure compliance with the institution’s dual use research policies.
- Maintain sufficient breadth of expertise to assess the dual use potential of the range of relevant life sciences research conducted at a given research facility.
- Maintain knowledge of dual use issues, concerns, and related institutional and Federal policies and understand risk assessment and risk management considerations. The review entity should be aware that a variety of risk mitigation measures are available and that designating research as DURC does not necessarily mean that the research should not be conducted or communicated.
- Make its procedures for reviewing life sciences research for dual use potential accessible to the public. The posted policies of the institution should include an overview of the institution’s procedures or review process, but should not include details of particular cases or the minutes of the DURC review entity’s proceedings.
- On a case by case basis, recuse any member of an institutional review entity who is involved in the research project in question or has a direct financial interest, except to provide specific information requested by the review entity.
- Engage in an ongoing dialogue with the PI of the research in question when developing appropriate risk mitigation plans.
- With the administrative infrastructure provided by the RPO, maintain records of institutional DURC reviews and completed risk mitigation plans for three years.
Each research initiative that is categorized as DURC is different and requires unique consideration, dependent on the information, materials, or technologies that may result from the research. Therefore, it is not possible to develop a single review process that can be used for all cases. The IRE performs its reviews based on the National Science Advisory Board for Biosecurity (NSABB) guidance.
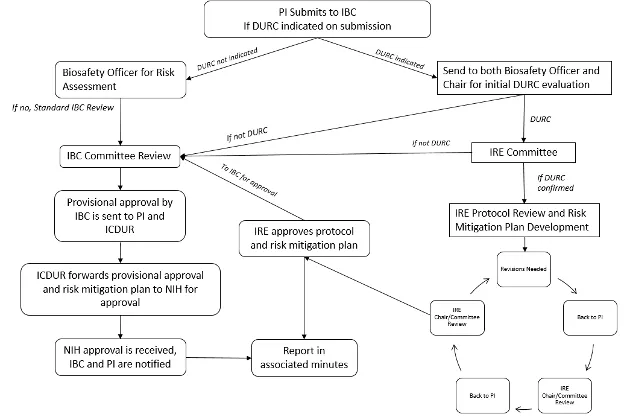
Minutes and Communication
The IRE committee maintains minutes of its deliberations and communicates its protocol review and risk mitigation plan recommendations in writing to the full IBC.
Appealing a Decision
Appeals may be made to the Vice President for Research through the ICDUR.
Post DURC Approval Requirements
Responsibilities of the Institution
- Within 30 calendar days of the institutional review of the research for DURC potential, notification of the Federal funding agency of any research that falls within the scope, including whether it meets or does not meet the definition of DURC. For non-Federally funded research, notification may be made to NIH (who may in turn notify the appropriate Federal funding agency, based upon the nature of the research); and
- Within 90 calendar days from the time that the institution determined the research to be DURC, provision of a copy of the risk mitigation plan to the funding agency for review – or for non-Federally funded research, provision of the plan to NIH for review (or referral to the appropriate funding agency).
- The ICDUR will provide all formal assurances to NIH and other Federal funding agencies about its DURC activity and compliance with the relevant policies in annual reports.
- The IBC follows its noncompliance reporting process ensuring that measures are undertaken by the institution to prevent recurrences of similar noncompliance. The noncompliance will be reported within 30 calendar days to the Federal funding agency or, for non-Federally funded research, to NIH (which will receive for administrative purposes on behalf of all of the institution’s Federal funders) and to the UVM Institutional Official.
Responsibilities of Principal Investigators
- Identify his or her research involving one or more of the DURC agents or toxins and follow the appropriate IBC and IRE review process for review of DURC potential.
- Work with the IRE to develop risk mitigation measures where appropriate.
- Conduct DURC in accordance with the provisions of the risk mitigation plan.
- Be knowledgeable about and comply with all institutional and Federal policies and requirements for oversight of DURC.
- Ensure that laboratory personnel conducting DURC (i.e., those under the supervision of laboratory leadership, including graduate students, postdoctoral fellows, research technicians, laboratory staff, and visiting scientists) have received education and training on DURC.
- Communicate DURC in a responsible manner. Communication of research and research findings is an essential activity for all researchers, and occurs throughout the research process, not simply at the point of publication. When researchers are planning to communicate DURC, it is their duty to ensure that it is done in a responsible manner, and in compliance with any risk mitigation plan approved by the IRE.
Education and Training
The IBC provides education and training on DURC for individuals conducting life sciences research that falls within the scope of this Policy. At a minimum, all DURC researchers will be required to complete the basic and DURC-specific module in CITI. Additional training is required consistent with the approved protocol and standard operating procedures. Links to educational materials developed by the Office of Biotechnology Activities is located on our IBC website.
The IBC maintains records of personnel training on dual use research for three years. As necessary, the IBC assists the PIs of life sciences research when questions arise about whether their research may require further review or oversight.
Risk Mitigation Plan
After a risk mitigation plan is developed by the PI and approved by the IRE, the research must be conducted in accordance with that plan and must be periodically reviewed by the institution to determine if additional modifications to the risk mitigation plan are appropriate.
Revised 03/15/19
4.0 Subsequent Submission Requirements
Once a Master Protocol Registration has been approved, the principal investigator has the responsibility of informing the IBC of changes in PI, key personnel, reporting incidents, submitting an annual continuing review, and closing the registration when the master protocol registration is no longer needed. Submissions are made to the IBC through the UVMClick system using the Amendment, Incident, Continuing Review, and Closure Request activities.
Revised 04/19/19
4.1 Amendment to Master Protocol Registration
Principal investigators revising a currently approved MPR must complete an Amendment smart form in UVMClick, revise appropriate smart form pages, and submit to the IBC for approval. Changes involving modification of biological agents, significant procedure changes (including change of the responsible principal investigator), changes to personnel, or changes that increase the risk of the project and/or the biosafety level must be approved by the IBC prior to implementing the changes.
Once approved, the Committee will return a signed approval memo back to the principal investigator.
NOTE: If the amendment involves vertebrate animals or human subjects, additional review by other committees may be required prior to implementation.
Revised 03/15/19
4.2 Addition of a New Funded Project to an Approved Master Protocol Registration
The IBC MPR is not grant specific and there is no need to submit multiple registrations for different funding sources. When a new grant is funded, an amendment to the existing MPR should be made to add the new source of funding and any new work involving biohazardous materials resulting from the approved grant. The amendment smart form should describe any new materials and any changes in research activities involving those materials. New or revised SOPs or BARDs must be submitted for review and approval as well.
Revised 03/15/19
4.3 Continuing Review of Master Protocol Registration
Annually, UVMClick-IBC will send a continuing review reminder to the principal investigator and his/her delegated proxy three months before the approval of an MPR is due to expire. Additional reminders will be sent at two months and one month prior to expiration. The principal investigator or proxy must log into the system and complete a continuing review smart form and submit to the Committee for review.
Once approved, the Committee will return a signed approval memo to the principal investigator.
Revised 03/15/19
4.4 Laboratory Accidents/Exposures/Spills (Incidents)
All biological exposures (i.e., life-threatening events), illnesses, or significant accidents leading to, or potentially leading to illness or that is environmentally dangerous to humans and/or animals must be reported to the IBC as soon as possible.
Initial review of the reported incident will take place through the IBC Policy and Procedure (P&P) Committee. This Committee includes a subset of current IBC members, including a member of the UVM biosafety team. Utilizing the P&P allows for a timelier review outside the monthly fully convened IBC meeting. The P&P Committee will review the information to assess the level of risk and whether suspension of work is required. Incidents that require reporting to the NIH OSP will be brought forward to the IBC at a regularly convened meeting at which time the IBC will be consulted regarding the level of significance and suggestions for additional safeguards or changes in procedures.
If applicable, additional research committee meetings will be convened to discuss the incident and all the biosafety procedures associated with the event. In some instances, the Chair of the IBC may suspend all relevant biohazardous materials in use by the PI pending a full investigation of the matter. Matters of noncompliance are separate from this procedure and are reviewed following Section 8 Procedures for Managing Noncompliance.
The IBC will share information about the reported incident, as applicable, with the Office of Animal Care Management, the Institutional Animal Care and Use Committee, and the Institutional Review Board.
The NIH Guidelines, under Section IV-B-2-b (7), specifically require UVM to report any significant problems with or violations of the NIH Guidelines, or any significant research-related accident or illness to the Institutional Official and NIH OSP within 30 days. Reports will be filed electronically to NIH OSP at NIHGuidelines@od.nih.gov.
Following recommendations from the IBC, the Institutional Official will inform external agencies such as the CDC, local public health department, State agencies, and funding sources about the incident and corrective actions.
Revised: 06/04/2025
4.5 Closing a Master Protocol Registration
In the event that the entire registration is closing, notification must be done by completing a Closure Request activity in UVMClick. This provides an opportunity for the researcher to update the Committee with any activities and describe the disposition of any remaining biohazardous materials. The closure request will be reviewed by the IBC and the researcher will be sent an official closure letter. The UVM biosafety team will be copied on the closure letter.
Revised: 7/24/24
4.6 Closure of Registration and Amendments by Committee
The IBC may require that an approved registration be closed in the following circumstances:
- If the work on a research protocol has not yet begun after a three-year period.
- If it is determined the principal investigator is no longer affiliated with UVM. In this instance, the Department Chair will be contacted to close the protocol or to assign a new PI.
- Closure may be required because of noncompliance. This would only occur after IBC review and communication with the investigator. Termination of IBC approval is reportable to the appropriate federal department or agency head and institutional officials.
In any of the situations described above, the IBC office will notify the PI, as well as his/her department chair, of the study closure.
Remove from Committee Consideration (registrations/amendments not yet approved)
The IBC may discard a new registration or amendment from continued consideration in the following circumstances:
- If the PI fails to respond to the IBC’s initial request for revisions and/or clarifications within 30 days post pre-review by the IBC Analyst (i.e., submission would be in clarification requested (pre-review) state.
- If the PI fails to respond to the IBC’s request for revisions and/or clarifications within 6 months post Committee review. (i.e., submission would be in the modifications required state.
Notification of Committee Closure or Removal of Protocol
In any of the situations described above, the IBC office will notify the PI, as well as their department chair, prior to study discard or closure.
Contact your IBC Analyst if sponsors, individuals, or processes outside of your control do not allow adherence to the response timeline criteria. Exceptions to response timeline criteria may be allowed on a case-by-case basis.
What If I Need to Reopen a Closed Protocol?
If an investigator needs to reopen a protocol after it has been formally closed with the IBC, the investigator would be required to submit a new protocol for review and approval.
Revised: 07/24/2024
5.0 Investigator Responsibilities
Investigators are responsible for complying with the IBC Policies and Procedures and any additional reporting requirements specified either by the sponsor or the regulating body. For example, any project that includes animals must be in compliance with the University’s Institutional Animal Care and Use Committee.
In addition, the principal investigator’s primary responsibilities include, but are not limited to the following:
Delegation of Responsibilities
PIs must personally perform or delegate to qualified co-investigators or research staff all the necessary tasks to carry out their studies. Even when specific tasks are delegated, the PI remains ultimately responsible for proper conduct of the study and fulfillment of all associated obligations.
Proxies are staff who are assigned by the PI to create, edit, and submit protocol materials on their behalf through the electronic protocol submission software. Proxies have the same control as the PI.
Proper Training and Oversight of the Research Team
The principal investigator is responsible for ensuring that the research team has appropriate training prior to and during the conduct of the study by:
- Reviewing with all laboratory staff the master protocol registration documents that describe the potential biohazards and the precautions to be taken (e.g., hazards and risks, immunizations, personal protective equipment required, decontamination, storage and disposal, spill procedures).
- Instructing staff in aseptic techniques and in the biology of the organisms used in the experiments so that the potential biohazards can be understood and appreciated.
- Faculty members, principal investigators and others responsible for directly, or indirectly, supervising labs will support and encourage a culture of safety and the use of best practices in the master protocol registration documents and procedures. This includes communicating safety and health as a core value, understanding the risks and requirements associated with the laboratories they oversee, assuring that appropriate precautions are taken against hazards and unsafe practices, that proper personal protective equipment is made available to all personnel, that workplace equipment and machinery is routinely maintained, that required medical surveillance of impacted employees is conducted, that regular safety inspections are performed and documented, and that students and employees receive job and hazard-specific safety training. (NOTE: This excerpt is taken from the UVM Laboratory Health and Safety Policy)
- Required web-based training: UVM has subscribed to the web-based training program, Collaborative Institutional Training Initiative (CITI). Personnel must complete the BSL-2 Basic Course in CITI and the Dual Use Research of Concern and Pathogens with Pandemic Potential.. Other required trainings as applicable to your master protocol registration, may include Animal Biosafety, Nanotechnology, or Select Agents. Web-based training is required and must be completed every three years. Reminder letters will be sent to personnel as their training expiration date nears. Reference the CITI Program Training page for additional information about required training and to check training completions. If working in BSL3 level containment, training requirements are described in Section 9.1 and 9.2. (NOTE: The IBC Committee will not approve key personnel until this requirement has been met.)
- Instructing and training laboratory staff in the practices and techniques required to ensure safety and the procedures for dealing with accidents.
- Informing laboratory staff of the reasons and provisions for any precautionary medical practices advised or requested.
- Supervising the safety performance of the laboratory staff to ensure that the required safety practices and techniques are employed.
- Investigating and reporting any significant problems pertaining to the operating and implementation of containment practices and procedures in writing to the IBC, NIH/OSP (as required), and/or other appropriate regulatory authorities.
- Correcting work errors and conditions that may result in the release of these materials.
- Ensuring the integrity of the biological and physical containment (biosafety level).
Adherence to Approved Projects
It is the principal investigator’s responsibility to conduct research in accordance with an IBC approved master protocol registration and ensure adherence at all times by the research team. This includes making sure that amendments and other follow-on submissions are submitted for IBC review in a timely manner, and then once approved, any changes are implemented by the research team.
Follow-on submissions include:
- Continuing Review reports
- Adverse events or incident reports
- Amendments regular or Amendments to add new grant funded projects
- Changes to research team
- Premature closure/suspension of activities
- Final study closure.
Access to Research Records
The investigator must provide direct access to all research records to the IBC staff. Research records refers to documentation of procedures and observations and other data pertinent to the research investigation. Dependent upon the project sponsorship there may be others with access needs, such as regulatory authorities.
Retention of Research Records
The regulatory mandated duration for records retention varies depending on which regulations apply to the research in question. UVM investigators need to ensure that their plan for record retention complies with the federal regulations as well as UVM Records Retention policy (PDF).
Revised 03/28/25
5.1 Study Team Member Responsibilities
Study team members are individuals carrying out research or teaching activities with biohazardous materials or an individual overseeing activities with biohazardous materials in an academic setting. Team members are responsible for:
Conducting activities in a safe manner and following laboratory procedures.
Reporting incidents to their supervisor/PI and/or the IBC.
Reviewing and understanding the Master Protocol Registration and its scope and safety requirements.
Completing applicable safety and laboratory specific training.
Acting as proxies on the PI’s behalf.
Revised 04/19/19
6.0 Conflict of Interest
As an academic research institution, we must continually dedicate ourselves to the integrity of the research enterprise.
IBC Members
All IBC members are required to disclose significant financial interests (SFI) in accordance with the University of Vermont Financial Conflict of Interest in Sponsored Research policy.
Selection of specific protocols for review by members is determined by the Committee's administrative staff and/or Chair. If an IBC member has a conflicting interest in a protocol (including, but not limited to being a principal investigator, a co-investigator, or a consultant on that protocol), that member may only provide information as requested by the IBC and will not be assigned to officially review nor vote on that protocol.
Investigators
Investigators are required to annually update their Significant Financial Conflict Interest Disclosure and complete training every 4 years through the CITI Program. More information regarding COI policy and procedures can be found on UVM’s Research Integrity site.
Revised 03/31/25
7.0 Post Approval Monitoring
To assist the UVM research community in adopting the best laboratory Biosafety practices that help ensure a safe working environment, the UVM Institutional Biosafety Committee has adopted the practice of regularly assessing laboratories associated with an active IBC registration. Assessments are an opportunity for laboratory personnel to receive guidance on prudent laboratory/Biosafety practices and procedures, ask questions and voice concerns. This process is in addition to regular site visits from a UVM Biosafety Team member.
Assessment Frequency
Assessment frequency will depend upon the level of risk associated with the laboratory work and the principal investigator’s compliance history. In the absence of any IBC related violations, UVM IBC will adhere to the following assessment schedule:
This schedule includes teaching laboratories.
BSL-1 laboratories – every 3 years
BSL-2 laboratories – every 2 years
BSL-3 laboratories – annually
The IBC has the discretion to request an off-cycle assessment when concerns arise involving complex procedures, investigators who are still in training, or compliance issues.
Process
A notice of visitation will be sent to the PI two weeks prior to the visit. RPO staff have been delegated authority by the Committee to conduct these visits, inviting IBC member(s) as needed. There are three potential outcomes:
- Visits without concerns will be documented in a report and signed off by RPO staff as completed.
- Visits with concerns or opportunities for improvement (no serious noncompliance) will be discussed with the PI and documented in a report to include any corrective actions. Corrective actions will be followed up at a subsequent visit as needed. Depending upon the findings, a draft report will be reviewed by Committee leadership for additional thoughts on corrective actions.
- Outcomes of a visit that include the potential for serious noncompliance will be managed following Policy Section 8. Procedures for Managing Noncompliance.
Updated: 3/31/25
8.0 Procedures for Managing Noncompliance
It is the responsibility of the Institutional Biosafety Committee (IBC) to address noncompliance with University policies and procedures. To exercise this authority, the IBC is empowered to inspect laboratories, procedure areas, animal housing areas, and to sequester research or training records. The IBC may receive reports via external complaints, internal complaints, Incident Reports, random and directed site visits with Biosafety Risk Assessments, and investigator or laboratory worker self-reporting. The IBC encourages faculty, staff and/or students to report instances of noncompliance.
This document describes the procedures for handling these matters. This policy is not all encompassing, and the IBC reserves the right to use its discretion in individual cases.
Definitions
Noncompliance is defined as the conduct of research in a manner that deviates from the approved protocol or disregards or violates federal regulations and/or institutional policies. Noncompliance may result from intended or inadvertent actions or omissions by study personnel, and can range from relatively minor or technical deviations to serious deviations that threaten the safety of personnel or the environment.
Serious Noncompliance is defined as noncompliance that, in the judgement of the IBC, potentially increases the risk of harm to personnel or the environment.
Continuing Noncompliance is defined as a pattern of noncompliance (recurring or ongoing) that, in the judgement of the IBC, may indicate an underlying deficiency in knowledge of the regulations or IBC requirements or an unwillingness or inability to comply with these regulations/requirements.
General Noncompliance Review Procedures
The investigation of potential noncompliance begins when the IBC becomes aware of potential noncompliance. This may include an allegation (unproved assertion) of noncompliance, a self-disclosure of noncompliance, or any other indication that noncompliance may have occurred. The process for the review of potential noncompliance involves initial administrative review, followed by an inquiry/fact finding process if indicated. Once complete, the IBC makes a determination as to whether the noncompliance is serious, continuing, or neither. The IBC determination will be documented in a summary report that contains a corrective action plan in cases of serious or continuing noncompliance. This process is detailed below, however at any point in the review process, the IBC Chair or Associate Chair, University Biosafety Officer, University Veterinarian, RPO Director or Assistant Directors, or another Institutional Representative designee may at their discretion:
- Recommend intervention for the safety of personnel or the environment
- Recommend the suspension of research activities
- Inform, involve, and/or provide salient documents to the PI, members of the research team, the Department Chair, Dean, Legal Counsel, or Institutional Officials, as appropriate
- Initiate reporting per federal regulations
- Initiate a monitoring visit
- Recommend immediate corrective actions
Process of Noncompliance Review and Determination
Initial Review of Allegation or Indication of Noncompliance: When there is an allegation or indication of noncompliance, the first step is an administrative review to determine if, in the judgement of the person(s) conducting the review, there is the potential for serious or continuing noncompliance. The initial review may be conducted by the RPO Director, RPO Assistant Director(s), an IBC Chair (Associate Chair or Chair), University Biosafety Officer, University Veterinarian or another Institutional Representative. Allegations/indications which are determined to have no potential to be serious and/or continuing noncompliance are resolved with either no follow-up (i.e. when an allegation or indication has no merit) or directly with the PI.
Inquiry/Fact Finding Process: If it is determined that the noncompliance has the potential to be serious or continuing or if questions remain following the initial review, then an inquiry (fact finding) process will begin. The particular circumstances of the noncompliance will determine when the fact finding begins and when the committee is briefed. The fact finding may be conducted by any IBC designee including a sub-committee or subcommittee member, the RPO Director, Assistant Director(s), an IBC Chair (Associate Chair or Chair) or other Institutional Representatives. The IBC may be briefed at any point throughout the fact finding process, as deemed appropriate by the designee. The fact finding process continues until the designee has arrived at a recommendation of determination (i.e. serious noncompliance and/or continuing noncompliance, or neither). A fact finding report is then prepared and includes the recommendation of determination and draft corrective actions. This fact finding report will be shared with the PI, and if applicable, other person(s) involved. All parties will be provided an opportunity to respond to any factual inaccuracies within the report before the committee deliberates.
Deliberation by the IBC: At a convened meeting, the IBC will consider all available information and make a determination as to whether the fact finding revealed serious noncompliance and/or continuing noncompliance, or neither. The following factors will be taken into consideration by the IBC or designee in making their initial determination as to whether the noncompliance is serious and/or continuing noncompliance. As each situation is unique, the indicators of noncompliance that are important in one case may not be relevant in other cases.
Factors in the Determination of Serious Noncompliance:
- Level of risk or potential risk to personnel or the environment
- Severity of safety violation
- Frequency or number of minor deviations or errors
- Intent
- Threat to integrity of the IBC review processes and requirements for the protection of personnel or the environment (i.e. falsification of IBC documents)
- Other factors that, in the judgement of the IBC or designee, are relevant to the situation being reviewed.
Factors in the Determination of Continuing Noncompliance: Similarity of noncompliance to previous deviations and/or noncompliance within the same registration or across registrations if the principal investigator has more than one registration. Likelihood that instances of noncompliance will continue without intervention
Final Determination of the IBC: If, in the judgement of the committee, the noncompliance is neither serious nor continuing, this determination will be shared with the PI. If, in the judgement of the committee, the noncompliance is serious and/or continuing. The designee will prepare a summary report including the IBC’s determination and an approved corrective action plan. This report will be shared with the PI, who will be given 7 days to review it before it becomes final.
Development of Corrective Action Plans:
The IBC/Biosafety Officer/designee will develop a proposed plan for corrective actions based on the information gathered during fact-finding and input from the principal investigator and/or other affected individuals. The proposed plan may:
- Require no further action
- Require minor corrective actions to achieve compliance
- Require additional education
- Require the investigator and/or other affected individuals to develop and implement procedures to prevent recurrence
- Review internal departmental or institutional mechanisms and systems for opportunities to prevent recurrence or similar occurrences by others
- Require additional oversight (e.g., by other faculty member or department process)
- Require more frequent IBC reviews
- Require internal monitoring visits or monitoring plans
- Suspend or terminate individual protocols
- Restrict researcher’s research activities
Requests for Reconsideration
A PI may request a reconsideration of the IBC’s determination. Requests must be limited to claims that either (1) the process was faulty, resulting in considerable risk that the outcome was incorrect; or (2) that the findings and/or corrective actions imposed by the IBC were excessive or unjustified. The written request must be submitted within 7 days of receipt of the summary report and must specify the nature of any claimed procedural error or the perceived unfairness of actions taken. Reconsiderations will be conducted by an IBC Chair (Chair or Associate Chair), Biosafety Officer, or Designee. The reconsideration process will result in one of three outcomes, either the summary report will stand and it will become final, the summary report will be modified and become final, or further investigation is necessary and will be initiated.
Required reporting
When noncompliance is determined to be serious and/or continuing, the final report will be forwarded to federal regulators as required, and to the Institutional Official, the Departmental Chair, the Dean, and sponsors, as applicable.
Guiding Principles for Noncompliance Review
Protection of Personnel and the Environment: The University of Vermont is committed to minimizing the risks to faculty, staff, students, the public, the facilities, and the environment while using biohazardous materials during research at UVM.
Fairness: The IBC strives to maintain a review that is impartial and honest, free from self-interest, prejudice or favoritism, including member recusal if such a self-reported or identified conflict arises.
Communication: The committee will communicate with the PI during the review process at points determined to be appropriate by the IBC designee.
Confidentiality: All IBC discussions and documents regarding a situation of noncompliance are considered sensitive and will be handled in a confidential manner and in accordance with state and federal regulations. The IBC cannot, however, guarantee complete anonymity to informants or witnesses. Confidentiality will be maintained to the extent possible to protect privacy and prevent retaliation, while still allowing for a full and fair review. Information may be shared, as described above under Required Reporting.
Conflict of Interest: Any IBC member who feels that they have a conflicting interest must recuse themselves from reviewing the issue of noncompliance. IBC members who are also listed as key personnel on the protocol(s) will not participate in the review but may be asked for information.
Procedures: In addition to what has been stated within this policy, the Committee will follow all applicable procedures that are outlined within the Policy and Procedures document.
Revised 03/31/2025
9.0 Animal/Biosafety Level 3 Facility
The University of Vermont’s Animal/Biosafety Level 3 (A/BSL-3) Facility is a high-containment core facility designed for both in vitro and in vivo research requiring containment at biosafety level 3 (BSL-3) and animal biosafety level 3 (A/BSL-3). Research conducted within the facility furthers the understanding and development of vaccines, diagnostics, and treatments of potentially life-threatening infectious diseases. The A/BSL-3 core is located within the Vermont Department of Health Laboratory (VDHL), adjacent to the UVM Colchester Research Facility (CRF). Use of these shared spaces is formalized in the VDH-UVM Joint Facility at Colchester Operations Agreement. Safe UVM operations in these facilities are formalized in the:
- UVM Biosafety Plan for the A/BSL-3 Facility
- UVM Incident Response Plan for the A/BSL-3 Facility
- UVM Security Plan for the A/BSL-3 Facility
The Vice President for Research (VPR) is the lead Institutional Official responsible for the assurance of research compliance in the biosafety area. The VPR has delegated oversight of operations of the A/BSL-3 Facility to the Research Protections Office (RPO) and Institutional Biosafety Committee (IBC). This core is supported by the Division of Safety and Compliance’s Environmental Health and Safety Department (EH&S), Office of Animal Care Management (OACM), and the Larner College of Medicine’s (LCOM) Senior Associate Dean for Research (SAD-R). Management authority is delegated in the Joint Agreement.
Responsibilities
The responsibilities for the appropriate conduct of research within this space are shared between UVM and VDHL. Below is an organizational chart along with the responsibilities of individuals or departments supporting the facility.

*UVM and VDH will each appoint 3 members to include BSO/RO, administrator, and one other member for a total of 6 members.
**UVM and VDH will each appoint 3 members of their choice who are affiliated with their institution for total of 6 members.
Institutional Biosafety Committee (IBC)
The UVM Institutional Biosafety Committee reviews all work with recombinant or synthetic nucleic acids in compliance with the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines). The IBC also reviews work with infectious agents and toxins of biological origin in compliance with the current Biosafety in Microbiological and Biomedical Laboratories (BMBL). The Committee’s A/BSL-3 responsibilities include:
- Reviewing and approving new and ongoing registrations including risk mitigation measures and procedures to be used.
- Advising core users on policies and procedures related to safe work at A/BSL-3 containment.
- Determining appropriate medical surveillance, as applicable, as part of protocol review.
- Advising UVM administration on the suspension of access privileges for staff found to be in violation of policies and procedures governing core use.
- The IBC Chair jointly leads the A/BSL-3 Users Group.
- IBC makes recommendations to the VPR regarding pertinent safety guidelines relating to procedures and facilities used in biological research.
- IBC is the gatekeeper for ensuring applicable training has been completed.
Vermont Department of Health Laboratory (VDHL)
The VDHL houses the A/BSL-3 facility and VDHL personnel are responsible for communicating to UVM:
- Any changes in VDHL policies or procedures.
- Scheduling of planned maintenance activities in coordination with UVM.
- Facility issues that may impact daily operations.
In addition:
- VDHL personnel grant access badges to UVM personnel for facility entry.
- VDHL personnel have representation on the A/BSL-3 Management and Operations Committees, as well as on the UVM IBC.
Joint A/BSL-3 Management Committee
The Joint A/BSL-3 Management Committee convenes to review all matters pertaining to the safety, security and scientific aspects of the protocols being carried out in the A/BSL-3 Facility, as well as the proper functioning of the A/BSL-3 Facility. The membership consists of three UVM and three VDHL employees. Issues that cannot be resolved at this committee level will be brought forth to the Joint A/BSL-3 Operations Committee for consideration.
Joint A/BSL-3 Operations Committee
The Joint A/BSL-3 Operations Committee is composed of three UVM and three VDHL employees and is charged with the development and review of administrative protocols relating to the VDHL/CRF complex, administrative oversight for the buildings and grounds, problem solving, and joint decision making.
UVM Larner College of Medicine
The UVM Larner College of Medicine (LCOM) appoints a member to the Joint Management Committee and resources the facility in the following ways:
BSL-3 Facility Manager
- Managing shared used calendar.
- Requesting PIN and badge access for prospective A/BSL-3 users upon completion of required trainings.
- Communicating time-sensitive issues to UVM and/or VDHL users and managing communications sent by VDHL and BGS.
- Updating and posting A/BSL-3 Facility signage.
- Recordkeeping of A/BSL-3 Facility maintenance and verification documents.
- Overseeing facility and equipment repairs, renovations, and maintenance.
- Providing coordinated support for escorting regulatory agency personnel, visitors, support personnel, contractors, and prospective new researchers through the facility.
- Collaborating with researchers, EH&S, OACM and VDHL to identify and resolve facility problems.
- Coordinating shipment of equipment and receipt of packages.
- Coordinating shutdown procedures and signing off on decontamination procedures.
Scientific Liaison
- Under the IBC, may function as senior mentor for new A/BSL-3 Principal Investigators (PIs).
- Subject matter expert in Research Plan Feasibility and Scientific Merit Review process.
- Provides oversight of new principal investigators to establish and/or verify proficiency along with the BSO.
- Facilitates communication among all stakeholders.
- Jointly leads the A/BSL-3 Users Group meetings.
Office of Animal Care Management (OACM)
OACM appoints a member of the Joint Management Committee and resources the facility in the following ways:
OACM Facility Manager
- Develop and maintain standard operating procedures specific to the ABSL-3 facility
- Conduct and/or delegate on an authorized user to train new staff on animal husbandry tasks, including decontamination and processing of used cages
- Maintain and ensure operations and readiness of the ABSL-3 facility and equipment including the cage wash, autoclave, biosafety cabinets, individually ventilated cage racks, cage-dumping station, etc.
- Provide or delegate hands-on animal handler training to research personnel
- Conduct proficiency assessments for OACM Husbandry Staff
- Prepare for and respond to incidents in the ABSL-3 facility
- On-call for emergencies
OACM Husbandry Staff
- Housekeeping in the animal holding rooms, anteroom, ABSL-3 corridor, and ABSL-3 decontamination room (i.e., researchers clean their individual animal procedure rooms), including safety tasks, except for those conducted by VDHL.
- Maintenance and inspection of the detergent room
- Management of biological waste in the ABSL-3 facility
- Advising researchers on animal welfare
- Prepare for and respond to incidents in the ABSL-3 facility.
- Report failure of equipment including cage wash, autoclave, biosafety cabinets, individually ventilated cage racks, cage-dumping station, etc.
OACM Veterinary Staff
- Provide weekday and weekend/holiday on-call veterinary services.
Environmental Health and Safety (EH&S)
EH&S appoints a member to the Joint Management Committee and resources the facility in the following ways:
UVM Biological Safety Officer (BSO)
BSO responsibilities can be found in the BMBL 6th edition. Below are some of the responsibilities:
- Reporting to the IBC and the institution any significant problems, violations of the NIH Guidelines, the BMBL, and any significant research-related incidents or illnesses.
- Providing technical advice to Principal Investigators and the IBC on research safety procedures.
- Serving as member on IBC.
- Advising mentors regarding new researcher training and conducting proficiency testing.
- Reviewing periodically the Biosafety, Security, and Incident Response Plans for the A/BSL-3 Facility.
- Conducting A/BSL-3 classroom training.
- Reviewing A/BSL-3 IBC protocols and conducting biosafety risk assessments.
- Designing and conducting drills and exercises.
- Providing technical input on facility renovation and safety equipment specifications.
Assistant Biosafety Officer (ABSO)
- Conducting periodic inspections to ensure that laboratory standards are rigorously followed.
- Assisting with the safe shipping of biohazardous materials and transportation between laboratories.
- Reviewing ABSL-3 IACUC protocols and conducting biosafety risk assessments.
- Serving as alternate member on IBC.
- Assuming the BSO’s duties in the absence of or as delegated by the BSO or EH&S Director.
Research Laboratory Personnel Responsibilities
Principal Investigator
- Ensure that all work is conducted in compliance with NIH, CDC, OSHA, and other applicable guidelines.
- Ensure lab is aware of and is following the Biosafety, Security, and Incident Response Plans for the A/BSL-3 Facility.
- Identify the initial risk level and periodically reassess the risk.
- Train, mentor and oversee the conduct of key personnel.
- Apply appropriate safety practices and procedures in the laboratories.
- Obtain prior IBC review and approval.
- Obtain prior IACUC review and approval as applicable.
- Ensure laboratory and equipment are in good working order.
- Ensure preventative maintenance and certification of equipment is conducted as required
- Attend A/BSL-3 Users Group meetings to discuss facility issues.
- Ensure all research personnel attend annual refresher trainings.
Study Team Members
- Ensure that all work is conducted in compliance with NIH, CDC, OSHA, and other applicable guidelines.
- Ensure they are familiar with Biosafety, Security, and Incident Response Plans for the A/BSL-3 Facility.
- Learn the operating procedures for the laboratory, the potential hazards of the infectious agents in use, and emergency procedures.
- Understand, through discussions with the PI and possible consultation with an occupational health provider, their personal limitations when working with these agents, report symptoms and/or illnesses suggestive of a laboratory-acquired infection (LAI), and any event that may result in an exposure or result in the creation of a potential hazard.
- Report any irregular laboratory conditions or accidents following the Incident Response Plan.
- Report any security issues following the Security Plan.
- Assist in the maintenance of the laboratory and equipment, including housekeeping and safety tasks.
- Assist in responding to incidents and accidents within the limits of their training and personal protective equipment.
A/BSL-3 User Group
The user group is comprised of A/BSL-3 researchers, laboratory staff, EH&S staff, OACM staff, IBC staff and LCOM support staff. The group is co-chaired by the IBC Chair and the scientific liaison. This group meets throughout the year to discuss facility issues, shutdown plans, and other items of interest to the users.
Research Plan Feasibility and Scientific Merit Review (for new users)
Unity of effort between the Principal Investigator, mentor, department chair, scientific liaison, designated college official (i.e., Senior Associate Dean for Research), and Office of the Vice President for Research (OVPR) ensures successful safety, training, and research outcomes. New investigators who are interested in using the A/BSL-3 core facility must complete the New Researcher Request to Use the A/BSL-3 Facility form. The completed form facilitates the following sequence of events:
- The Principal Investigator expresses A/BSL-3 research interest to his/her research mentor and department chair. The Principal Investigator must demonstrate a research history leading to a logical evolution to A/BSL-3 research (i.e., education, training, funding history). Institutional technical expertise requirements are below.
- The Department Chair must assess research potential, necessary investment and training, and the Principal Investigator’s ability to meet A/BSL-3 core facility use criteria.
- The Department Chair should consult with the LCOM Senior Associate Dean for Research as part of the decision-making process.
- The IBC Scientific Liaison will provide a review/recommendation summary to the respective designated college official to either: 1) endorse the PI’s research plan for submission to the IBC for review, or b) provide actionable feedback to the PI to address next steps. The designated college official will then review those research projects that are endorsed by the Department Chair and IBC Scientific Liaison to assess the proposed facility use and how the research fits into LCOM’s and UVM’s overall strategic plan.
- If the new researcher has been approved to use the facility through this process, the research project may be submitted to the IBC for review. As part of the submission, the PI must identify a qualified A/BSL-3 research PI (scientific mentor).
Technical Expertise Requirement
Researchers wishing to utilize the A/BSL-3 facility must meet the minimum prior experience criteria as outlined in Section 9.1 Biosafety Level 3 Training Guidelines and Practices for Researchers.
Pre-Entry Training Program
Each researcher will be required to enter a training and mentoring program based upon their prior experience. See Section 9.1 Animal/Biosafety Level 3 (A/BSL-3) Training Guidelines and Practices for Researchers for further information.
Animal husbandry staff are required to complete a set of training requirements specific to their tasks prior to entering the facility. See Section 9.2 Animal/Biosafety Level 3 (A/BSL-3) Training Guidelines and Practices for Animal Care Staff for additional information.
Access to the Core Facility
Use of the A/BSL-3 facility is restricted to those researchers, animal care staff, and Biosafety
Team members approved for entry by the Institutional Biosafety Committee. New study team members will be provided with IBC provisional approval to access the space escorted for necessary training. Once training is complete, the IBC will review the training and proficiency documentation and provide IBC approval for independent access. Following this final approval by the IBC, researchers and animal care staff will complete the A/BSL-3 Facility Tour with VDHL’s PH Lab Safety Compliance Chief to gain independent access to the facility.
Submitting to the IBC
Registration for a project that involves Risk Group 3 agents requires prior IBC review and approval. This review will be conducted by the Full Committee. The submission requirements are the same as submissions for BSL-2 research projects and can be found on the IBC webpage.
Revised: 06/03/25
9.1 Biosafety Level 3 (A/BSL-3) Training Guidelines and Practices for Researchers
Background Information
Animal/Biosafety Level 3 (A/BSL-3) is applicable to clinical, diagnostic, teaching, research, or production facilities where work is performed with indigenous or exotic agents that may cause serious or potentially lethal disease through the inhalation route of exposure. Laboratory researchers and their research personnel must receive specific training in handling pathogenic and potentially lethal agents and must be supervised by scientists competent in handling infectious agents and associated procedures. The A/BSL-3 laboratory has special engineering and design features.
Training of researchers to work in the A/BSL-3 Facility must conform to the:
- UVM Biosafety Plan for the A/BSL-3 Facility
- UVM Incident Response Plan for the A/BSL-3 Facility
- UVM Security Plan for the A/BSL-3 Facility
The minimum requirements for a Principal Investigator to register an A/BSL-3 research program are:
- A terminal degree (PhD and/or MD, DVM)
- Two years of practical hands-on or oversight experience working at BSL-2 or ABSL-2 labs with human pathogens withinthe last 5 years
- Nomination by Department Chair (provided through the Researcher Request to Use A/BSL-3 Facility Form[DS1])
- Identifying and obtaining training commitment from an A/BSL-3 scientific mentor
- Completion of A/BSL-3 research training program and proficiency testing
- Establishment of an approved research training plan for trainees including laboratory staff and predoctoral trainees, postdoctoral trainees, and other faculty.
The minimum requirements for non-PI researchers to participate in an A/BSL-3 certificationprogram are:
- Bachelor’s degree in applicable scientific discipline
- Nomination by Principal Investigator (provided through the Researcher Request to Use A/BSL-3 Facility Form)[DS2] [DS3]
Two years of practical hands-on experience workingat BSL-2 or A/BSL-2 labs within the last 5 years is preferred for all A/BSL-3 researchers.
UVM research personnel must complete the trainingplans outlined below to be authorized to work in the A/BSL-3 facility located in the Vermont Department of Health Laboratory (VDHL) in Colchester, Vermont. Allpersonnel working in the A/BSL-3 facility must adhere to all UVM and VDHL A/BSL-3 Standard Operating Procedures (SOPs). These may include following federal regulations for CDC Select Agents andToxins if these agents are in usein the facility by VDHL personnel. UVM personnel working with animals at AnimalBiosafety Level 3 must complete additional training, which is agent-specific, as outlined below.
Roles
Mentor – an individual who has previously completed the training process and has been working independently in an A/BSL-3 facility for at least 2 years. This may be the PI of a registration, laboratory personnel, or an approved external body.
Trainee – individual undergoing the training process to obtain access to the A/BSL-3 facility.
Animal Care Personnel – Animal husbandry staff and veterinary support staff requiring facility access to perform job duties involving housekeeping, cage changing, animal welfare monitoring and provision of veterinary care.
Scope of A/BSL-3 Training
All research personnel must complete steps #1-18 as outlined in the Official Training Record for New A/BSL-3 Users for researchers new to the UVM A/BSL-3 facility. Training includes a mixture of online and in-person classroom/didactic training, orientation to the facility, and practical, hands-on training with a mentoring period.
Animal care personnel will be provided with general and facility-specific training, as outlined in Section 9.2 Animal/Biosafety Level 3 (A/BSL-3) Training Requirements for Animal Care Staff.
Mentorship Procedures
Because experience and training can be accomplished at different rates, mentorship periods willvary in length for each trainee. Ideally, the mentor will be a UVM faculty member, researcher, or senior staff.
If independent access is not granted after 12 months, trainees may continue to work under mentored supervision in A/BSL-3 laboratories. Trainees may also work at A/BSL-2 for an additional year and then requestadditional training to begranted independent access for the A/BSL-3 facility.Mentors and the UVM BSO willconsult with the PIs to determine the bestsolution on a case-by-case basis.
Principles of Mentored A/BSL-3 Training
The Principal Investigator (PI) of an A/BSL-3 registration is ultimately responsible for ensuring that research personnel have received the appropriate training by an approved A/BSL-3 mentor. The A/BSL-3 mentor may be the PI or an experienced, authorized A/BSL-3 facility user with full access, assigned by the PI and approved by the UVM IBC. The mentor must have at least 2 years of experience working with RG 3 agents in an A/BSL-3 laboratory. It is also recommended that the mentor have hands-on experience commensurate with the type of experiments that will be conducted by the trainee. A mentor may be assigned from the PI’s own laboratory or be obtained from another internal or external resource (e.g., different research group, external A/BSL-3 contractor).
The UVM BSO participates in the initial orientation phase and the final validation phase of the training program as described below. The BSO is also available for consultation during the mentoring process.
A/BSL-3 Training Requirements
The guiding principles and metrics of mentored training (i.e., steps #13 and #14 on the training record form found in the forms section) are specified below.
Checklist Sections 13 – 14 outlining steps
13. Complete A/BSL-3 orientation and general training with mentor and/or Biosafety Team member
- Entry 1 (Donning/Doffing and Entry/Exit
- Entry 2 (Biological Spill and Emergency Responses)
- Entry 3 (Experimental SOPs and Specific Work Practices)
- There are two different training tracks.
- General Laboratory Research (In Vitro): This track is designated for personnel that will conduct research with agents inthe in vitro BSL-3 suites only.
- Animal Research (In Vivo): This track is designated for personnel that will conduct experiments or assist withanimal procedures at ABSL-3only.
- If researchers are conducting both in vitro and in vivo work, both tracks are required.
14a. Mentored rehearsal of A/BSL-3 procedures in an A/BSL-2 laboratory
Option 1: Locally mentored (supervised) training
- Personnel willfollow A/BSL-3 practices and procedures but work with an RG 2 agent.
- The number of directly supervised hours is dependent upon the individual’s combined experience and demonstrated competency and is solely at the discretion of the mentor and BSO. Disagreement between the mentor and BSO will be adjudicated through the IBC.
- Qualifications for mentors:
- Minimum of 2 years of A/BSL-3 experience.
- Experienced with the specific agent the trainee will be studying or with other RG-3 agent(s) requiring similar biosafety work practices.
- Approved to work independently in the A/BSL-3 facility.
- Must be full-time faculty, researcher, or senior staff.
- The PI must submit an amendment to the IBC registration through UVMClick to add this new trainee and propose a mentor. Therequest must include a section describing all the above qualifications of the mentor. IBC approval may be granted through the IBC designated review process to allow access to the A/BSL-3 facility in a training capacity.
Option 2: Externally mentored (supervised) training
- In rare instances where a UVM affiliated mentor is unavailable, a non-UVM affiliated mentor may be used for A/BSL-3 training either on-site at UVM or off-site at another A/BSL-3 institution.
- PI (or perhaps Department, LCOM) would fund the training.
- Proposed external A/BSL-3 training must be reviewed and approved by the Policies and Procedures Subgroup of the IBC.
- A UVM facility orientation will occur with the trainee in accordance with existing training process prior to the external training.
- Certification/completion of training from the external trainers will be required.
- This certification must include the completion of the attached BSL-3 and/or ABSL-3 training checklists.
- An independent proficiency assessment of A/BSL-3 work practices will be completed by the UVM BSO prior to granting access.
As an example, the Ragon Institute of MGH, MIT and Harvard provides BSL-3 training in a fee-for-service model. The portfolio of training includes the technical execution of experimental procedures.
14b. Initial proficiency evaluation at A/BSL-2
- Trainees will beevaluated by their approved mentor with microbiologicaltechniques and procedures as applicable.
- External mentors will be required to provide certification of competency for the trainee.
- An evaluation will be performed to identify competence in the area described in the Techniques Evaluation Worksheet (Form A).[DS4] [DS5]
- Once found competent, the trainee will move onto mentored training within the A/BSL-3 facility.
14c. Mentored training of research/work practices at A/BSL-3 for General Laboratory Research & Animal Research
Trainees must be mentored and always directly supervised by an IBC approvedmentor until they areproficient in all A/BSL-3 practices and procedures in the A/BSL-3 facility.
- The mentoring period willvary for each trainee andwill be determined by the mentor and BSO (typically 15-30 hours for individuals with some experience and typically 30-60 hours for individuals with no previous experience in A/BSL-3). The number of directly supervised hours is dependent upon the individual’s combined experience and demonstrated competency and is solely at the discretion of the mentor and BSO. Disagreement between the mentor and BSO will be adjudicated through the IBC.
- Training verificationwill be documented by the mentor by completion of the A/BSL-3 Training Proficiency Certification Checklist (Form B). [DS6] [DS7] The trainee must demonstrate proficiency and competence in all relevant areas specified on the checklist.
- The mentor will submit the completed checklist to the PI (if not the mentor) and UVM BSO.
14d. Secondary proficiency assessment of A/BSL-3 work practices by the Biosafety Team
- The Biosafety Team will independently assess the proficiency of the trainee in each of the areas specified in the Training Proficiency Certification Checklist (Form B).
- The Biosafety Team member will complete his/her section of the attached training proficiency checklist to certify the trainee has demonstrated competency in eachof the areas specified in the training checklist.
- The UVM Biosafety Team member willcoordinate with the VDHL BSO to complete final VDHL training and approve independent access.
The PI will submit the completed certification of competency to the IBC, who will approve independent access.
| A/BSL-3 Required Training Flow | |
|---|---|
| Candidate must request facility badge and pin access from the Vermont Department of Health | |
| ↓ UVM Internal Training ↓ | ↓ External Training ↓ |
| Mentor for the candidate must be identified — experience requirements for both mentor and candidate listed above | External mentor (must provide credentials and training plan) and prior approval is required of the IBC |
| Orientation to A/BSL-3 by mentor or biosafety team member | Orientation to UVM A/BSL-3 facility by UVM biosafety team member |
| Mentored training at BSL-2 | IBC approved training plan is carried out and UVM training record and proficiency are provided to the IBC |
| Proficiency determined by mentor or biosafety team member | Reorientation and proficiency evaluation with scientific liaison and biosafety team member |
| Mentored training in A/BSL-3 | |
| Proficiency evaluation by mentor and biosafety team member | |
Revised: 06/03/25
9.2 Biosafety Level 3 (A/BSL-3) Training Guidelines and Practices for Animal Care Staff
Animal care personnel will be provided with general and facility-specific training, as well as applicable husbandry and animal care training, by the OACM Facility Manager, an animal care technician or UVM Veterinarian who has completed the training and has been granted independent access to the facility. These roles would be considered the mentor.
Prior Experience
Animal care personnel will have 3-6 months prior experience at ABSL-2.
Initial A/BSL-3 Training
Prerequisites
- Complete online CITI Animal Biosafety Course.
- Candidates must enroll in the Occupational Health Program and receive medical clearance to wear a respirator (PAPR) and work in the ABSL-3 Facility.
- OACM Facility Manager will file a request for facility badge and pin access from VDHL.
Orientation
Orientation includes review and discussion of the following facility plans with the mentor:
- UVM Biosafety Plan for the A/BSL-3 Facility
- UVM Incident Response Plan for the A/BSL-3 Facility
- UVM Security Plan for the A/BSL-3 Facility
A review of the A/BSL-3 Facility Standard Operating Procedures (SOPs) will also take place.
Practice at A/BSL-2
The mentor will work with the candidate on performing the items from the Plans and SOPs at ABSL-2 until competency is achieved. This work will include, but is not limited to, the following:
- Entry and exit procedures
- Building
- Facility
- Donning/doffing PPE as applicable
- Animal handling
- Bringing animals into ABSL-3
- Cage change in BSC
- Handling and disposal of soiled cage bedding
- Disposal of carcasses
- Biosafety cabinet (BSC) use
- Preparation and decontamination of BSC
- Cage change in BSC – buddy system and single person
- Waste management
- Autoclave operation for each type of biohazard waste
- Chemical waste
- Incident/Emergency
- Emergency contact list
- Spills inside BSC
- Spills outside BSC
- Animal escapee
- General facility failures
- Critical system alarms (e.g. HVAC, freezer, etc.)
- Medical emergencies
- Security issues
- Case scenarios; incidents during business hours vs. after hours
Practice and Competency Inside the ABSL-3 Facility
After provisional IBC approval is received, trainees will be escorted into the facility with their mentor. Their mentor will observe and correct, where applicable, those tasks previously practiced at A/BSL-2. Once the trainee has demonstrated proficiency conducting these tasks within the A/BSL-3 facility, they will be recommended for full access.
Training and Proficiency Records
Training and proficiency details will be documented on the Training Record for Animal Care Staff Form C and be maintained by the OACM Facility Manager. OACM may tailor the checklist to meet the needs of the position; for instance, vet techs and veterinarians may not need to be assessed for cage changing.
Ongoing Training and Educational Opportunities
As new agents are introduced to the facility, animal care staff are updated by the researcher, OACM Facility Manager, and/or Biosafety Team member with the following information:
- Agent-specific safety data sheet (PSDS)
- Specific animal handling precautions
- Specific decontamination procedures
The OACM Facility Manager will ensure animal care staff are afforded opportunities to take part in professional development and participate in facility educational opportunities, required drills, etc. Refresher hands-on training will be repeated as necessary. The OACM Facility Manager is responsible for confirming that the staff are up to date with the following training:
- Bloodborne pathogen
- Lab safety
- IIPP
- Waste management, and
- ABSL-3 medical clearance
Revised: 6/12/25