- ancestors of organisms
- phylogenetic trees
- protein structures
- protein function
Biocomputing in a Nutshell
Ulf Reimer and Georg Fuellen 
A Short View onto the Development of Biology
The success of modern molecular biology might be considered a cartesian dream. Reductionism, Rene Descartes' belief of understanding complex phenomena by reducing them to their constituent parts - despite all its limitations - has turned out to be a home run in molecular biology.The developments in modern biology have their roots in the interdisciplinary work of scientists from many fields. This was a crucial element in the breaking of the code of life; Max Delbrück, Francis Crick and Maurice Wilkins all had backgrounds in physics. In fact, it was the physicist Erwin Schrödinger (ever heard about Schrödinger's cat ?), who in "What is life" was the first to suggest that the "gene" could be viewed as an information carrier whose physical structure corresponds to a succession of elements in a hereditary code script. This later turned out to be the DNA, one of the two types of molecules "on which life is built".
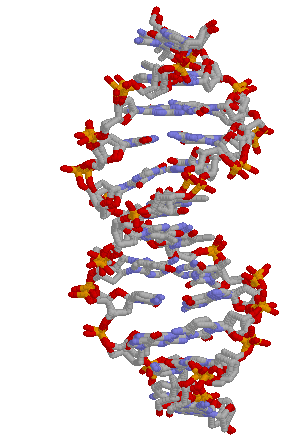
The 3-dimensional structure of the DNA. Our
homepage
features an animation of DNA, and our background
image is based on it.
{kind=link}
Linus Pauling, the chemist, vitamin C-ist and anti atom-bombist determined the structure of the other type of molecule, the protein molecule - that is chains made up of things called amino acids.
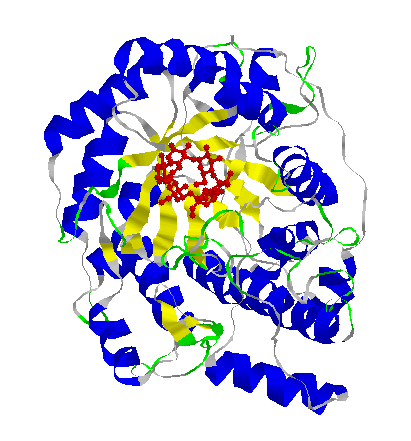
The 3-dimensional structure of a protein,
Beta-amylase. The main structural units of the protein, which are made
up of just a few amino acids each, are differently coloured.
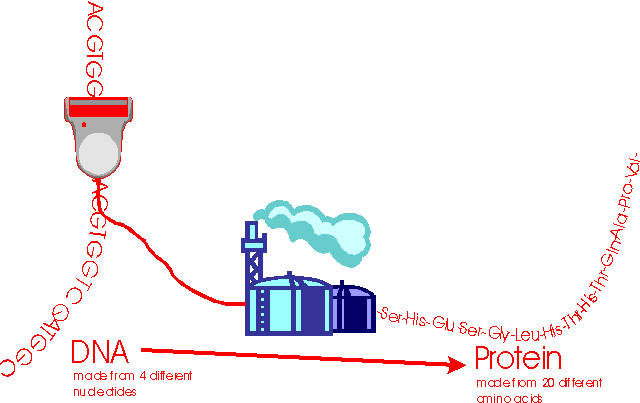
This work inspired James Watson and Francis Crick in 1953 to elucidate the structure of DNA - the ABC of all known living matter. To cut a long story short over the next years many people pieced the puzzle together: The building blocks of life are the 20 amino acids that make up proteins; DNA contains the blueprints for these structures in its own structure. It is a long strand made of 4 nucleotides - this is the code of life. It goes ACGTTCCTCCCGGGCTCC, and so on, and so on, and so on. If you know the code you know the structure of all living things, at least in theory.
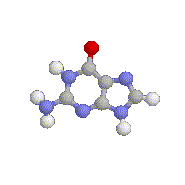
An animation of Guanine (G), one of the
4
standard nucleotide bases. The colored balls represent the atoms from
which it is made. Similar ball-and-stick models can be constructed for
the 20
amino acids. (Click here
if you'd like to `animate' the Guanine.)
Here is a summary of the relationship between DNA and protein:
An Enourmous Flood of Data
Restless technology has produced means of reading genes (DNA) almost like bar - code. The problem is that life is a complicated business, and therefore the code to describe even the smallest of God's creatures would fill many books. But scientists are very ambitious people and do lots of over-time. They have started to decode "themselves" in the Human Genome Project - HUGO for short. In fact, a sort of "average" human is decoded sampling DNA from unknown donors. But the difference in DNA between any human, and another one (or a scientist...) is almost null. Nevertheless, an average human scientist is made up of about 2.9 billion (2.9*109) nucleotides !This orgy of reductionism presents problems which only big brother can solve: How do I store all this information in a form which is universally accessible and retrievable? What started as a cartesian dream is turning out to Bill Gates' satisfaction: Computers are needed !
Vast computer data banks accessible to you and me store this vast quantity of information. There are a lot of different data banks where DNA and protein sequence information are stored. Three examples are listed in the table below.
| Name of data bank | Type of sequences stored | Number of sequences (1996) |
|---|---|---|
| EMBL / GENBANK | Nucleotide sequences | 827174 |
| SWISSPROT | Protein sequences | 52205 |
| PDB | Protein structures | 4525 |
The growth of one typical data bank is shown in below, the increasing number of sequences in the SWISSPROT data bank as time goes by.
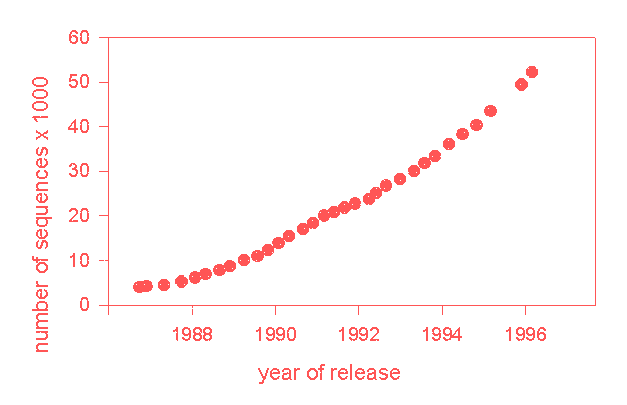
Growth of the SWISSPROT data bank.
How can We Analyze the Flood of Data ?
An advantage of these data banks is their flexibility. All this information can be ordered and combined according to different patterns and tell us an awful lot.The motto goes: don't just store it, analyze it ! By comparing sequences, one can find out about things like
Phylogenetic trees are genealogical trees which are built up with information gained from the comparison of the amino acid sequences of a protein like cytochrome C, sampled from different species. Proteins like Beta-amylase or Hemoglobin cannot be chosen to get the "full picture", that is the full tree, because they don't occur throughout the living matter. Due to Darwinian Evolution, the protein has a slightly different amino acid sequence for each of the species. One phylogenetic tree was created for instance with the sequences of cytochrome C from several plants, animals and fungi. Below, part of this phylogenetic tree is shown.
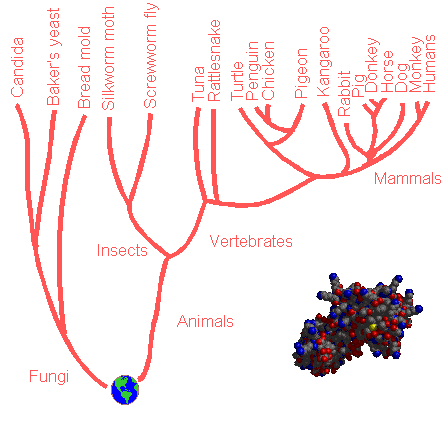
Drawing of a phylogenetic tree based on the
amino acid sequence data of cytocrome
C (see inset).
{kind=link}
Prediction of protein structure from sequence is one of the most challenging tasks in today's computational biology. More or less, the task is to calculate an image like the one in the second figure of this text. Although most information of 3-dimensional structure is encoded in the amino acid sequence it is still unknown which information controls the process of protein folding. Among millions of possible folding products, proteins take up one working, native structure. Since it is very difficult and expensive to evaluate structures by methods like X-ray diffraction or NMR spectroscopy, there is a big need for the unfailing prediction of 3-dimensional structures of proteins from sequence data. Today there are methods which are able to give a quite reliable result from available sequence data, the odds to get this "right" are about 65%.
Sequence comparison is a very powerful tool in molecular biology,
genetics and protein chemistry. Frequently it is unknown for which proteins
a new DNA sequence codes or if it codes for any protein at all. If you
compare a new coding sequence with all known sequences there is a high
probability to find a similiar sequence. Often it is already known which
role the protein in the data bank plays in the cell. If you assume that
a similar sequence implies a similar function, you now have much more knowledge
about your new sequence than before. (See also the contribution
by Joelle Thonnard in this volume.)
Proteins of one class often show a few amino acids that always occur
at the same positions in the amino acid sequence. By looking for "patterns"
you will be able to gain information about the activity of a protein of
which only the gene (DNA) is known. Evaluation of such patterns yields
information about the architecture of proteins. Often these patterns are
involved in active sites, which are the workbenchs of proteins.
What is our task in this field ?
A lot of complicated algorithms have been created. There are tools to scan data banks for sequences as FASTA and BLAST are. There are programs like Clustal and MSA for comparing sequences. There are hundreds more. Although the development of new tools is more transparent because of the possibilities of the Internet, it is not easy to keep up with everything. Exploitation of these possibilities requires a new breed of scientist: those versed in information technology AND biology, and they may enable us go where no man has gone before. Through a new surge of interdisciplinarity it may be possible to transcend the limits of reductionism; from the vast quantities of bytes and pieces, the contours of complex structures and relationships might emerge from the genetic alphabet soup as life itself once emerged from the primordial soup.To fullfill this promise there are now interdisciplinary Internet courses where people can learn about biocomputing...
Ulf Reimer and Georg Fuellen
Back to Biocomputing For Everyone WWW Pages