Our
Research
I. Fundamental studies on the reactivity of 1-aza-2-azoniaallene salts leads to preparative methods for a wide-range of nitrogen heterocycles:
My research group has been studying
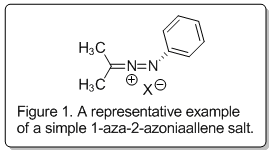
1-aza-2-azoniaallene salts (Figure 1) in the context of intramolecular
reactions for several years now. These studies have led us to discover
that these high-energy heteroallene intermediates can react by multiple
mechanistically-distinct pathways to afford structurally diverse
heterocyclic scaffolds. This is important from a fundamental reactivity
standpoint (we have shown that these intermediates can react in
unprecedented ways), and also from a practical application standpoint
(the products formed are structurally unique and could be useful either
for biological studies or as synthetic intermediates).
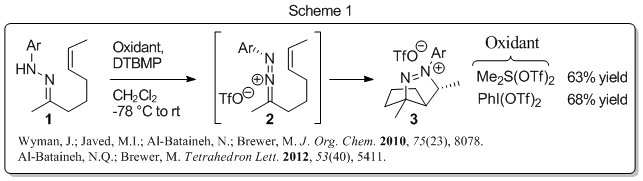
When we began this work, little was known about the reactivity of 1-aza-2-azoniaallene salts save that they were highly electrophilic on carbon (nucleophiles readily add at that position), and that they could participate in (3 + 2) cycloaddition reactions with unsaturated partners. Our work initially focused on studying intramolecular (3 + 2) cycloadditions of 1-aza-2-azoniaallene salts with alkenes. We observed that this is a fairly general reaction that provides bicyclic diazenium salts (e.g. 3, Scheme 1) in good yield, and we established new procedures for preparing the reactive 1-aza-2-azoniaallene salts in situ from hydrazone precursors via chemical oxidation.
We have since discovered that
1-aza-2-azoniaallene salts can react by several alternative pathways.
For example, by changing the length of the tether that connects the
reacting functional groups, the (3 + 2) cycloaddition becomes
unfavorable and an unprecedented (4 + 2) cycloaddition instead occurs to
give a tricyclic protonated azomethine imine containing a
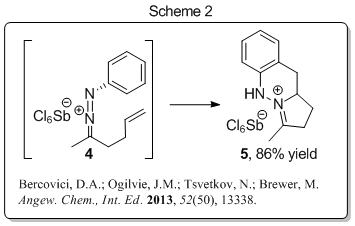
1,2,3,4-tetrahydrocinnoline scaffold (Scheme 2, 4 to
5). Our experimental studies
have shown that this reaction is quite general with respect to alkene
substitution and delivers products that contain all carbon or nitrogen
bearing quaternary centers in high yield. Our experimental results also
showed that this reaction is stereospecific with respect to alkene
geometry, and thus most likely bonding concerted. The discovery of this
reaction is important because it brings to light an unknown reactivity
of these heteroallene intermediates, and the products are unique and
non-trivial and will allow biomedical researchers to explore new
chemical space. In addition, the products formed by this reaction are
themselves functional group rich compounds that could be used as
synthetic intermediates. We are now in the process of applying this
methodology as a key step in the total synthesis of the
naturally-occurring indole alkaloid ibophyllidine.
Importantly,
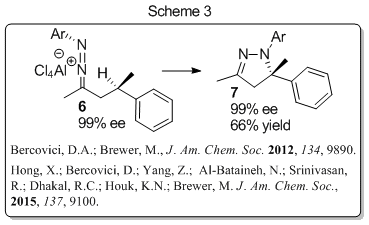
we have also discovered that 1-aza-2-azoniaallene salts can participate
in C-H insertion reactions to form a new C-N bond in what is known as a
C-H amination reaction (Scheme 3). Reactions that result in the direct
functionalization of unactivated C–H bonds are powerful transformations
because they provide new synthetic strategies that can be more efficient
and simpler to execute than traditional synthetic sequences. C–H
amination reactions are important because they allow for the selective
installation of nitrogen atoms at a late stage in a synthetic sequence
and provide fundamentally new approaches to the preparation of nitrogen
heterocycles. In our case, the products formed are pyrazolines, which
are important motifs in many biologically active compounds. Insertion
occurs at benzylic, allylic, and tertiary aliphatic centers to provide a
wide range of pyrazoline or pyrazole products.
We have studied the cycloaddition and insertion reactivity of 1-aza-2-azoniaallene salts computationally in collaboration with Prof. K. N. Houk (UCLA). These studies determined how tethers control whether an intramolecular (3+2) or (4+2) cycloaddition occurs between an aryl-1-aza-2-azoniaallene salt and a pendent alkene. While the (3+2) cycloaddition is intrinsically more favorable than the competing (4+2) cycloaddition, shortening the tether significantly destabilizes the (3+2) cycloaddition transition state because of unfavorable interactions within the forming four-membered ring. On the other hand, the shorter tether stabilizes the (4+2) transition state and facilitates this novel transformation. Perhaps more importantly, these studies also supported our assertion that the (4+2) cycloaddition occurs by a concerted Diels-Alder-like transition state.
Our computational studies of how 1-aza-2-azoniaallene salts insert into C–H bonds gave very interesting results. These studies show that C-H amination proceeds through a hydride transfer transition state to form a N-H bond and a transient carbocation, and then C-N bond formation occurs to generate the heterocyclic product. The intrinsic reaction coordinate diagram for this insertion process shows that the two consecutive bond formation processes (N-H and C-N) are energetically concerted and the second step (C-N bond formation) occurs much faster than C-C bond rotation, which explains our experimental observation that the C-H amination occurs at a chiral center without stereomutation. The results of these mechanistic studies are important because this bonding-stepwise mechanism is unique to this insertion reaction (all other aminations occur through radical pathways or concerted process in which both N–H and N–C bonds form at the same time) and it has made us appreciate that these species are a special form of a nitrenium ion – specifically an imino-nitrenium cation. With this in mind, we have begun evaluating other reactions that imino-nitrenium cations might undergo.
Related Publications:
Hong, X.; Bercovici, D.; Yang, Z.; Al-Bataineh, N.; Srinivasan, R.; Dhakal, R.; Houk, K.N.; Brewer, M. "Mechanism and Dynamics of Intramolecular C-H Insertion Reactions of 1-Aza-2-azoniaallene Salts" Journal of the American Chemical Society, 2015, 137, 9100-9107.
Hong, X.; Liang, Y.; Brewer, M.; Houk, K.N. “How Tethers Control the Chemo- and Regio-Selectivities of Intramolecular Cycloadditions between Aryl-1-Aza-2-Azoniaallenes and Alkenes” Organic Letters 2014, 16(16), 4260-4263. (DOI: 10.1021/ol501958s)
Bercovici, D.A.; Ogilvie, J.M.; Tsvetkov, N.; Brewer, M. “Intramolecular Polar [4Å + 2]-Cycloadditions of Aryl-1-aza-2-azoniaallene Salts: Unprecedented Reactivity Leading to Polycyclic Protonated Azomethine Imines” Angewandte Chemie, International Edition, 2013, 52(50), 13338-13341. (DOI: 10.1002/anie.201306553)
Al Bataineh, N.Q.; Brewer, M. “ Iodine(III)-mediated bicyclic diazenium salt formation”, Tetrahedron Letters, 2012, 53, 5411-5413. (DOI: 10.1016/j.tetlet.2012.07.116)
Bercovici, D.A.; Brewer, M. “Stereospecific intramolecular C–H amination of 1–aza–2–azoniaallene salts”, Journal of the American Chemical Society, 2012, 134(24) 9890-9893. (DOI: 10.1021/ja303054c)
Wyman, J.; Javed, M.I.; Al-Betaineh, N.; Brewer, M. “Synthetic Approaches to Bicyclic Diazenium Salts”, The Journal of Organic Chemistry, 2010, 75(23), 8078-8087. (DOI: 10.1021/jo101706h)
Javed, M.I.; Wyman, J.M.; Brewer, M. “Synthesis of Fused and Bridged Bicyclic Diazenium Salts by Intramolecular Cycloaddition”, Organic Letters, 2009, 11(10), 2189–2192. (DOI: 10.1021/ol900502s)
Wyman, J.M.; Jochum, S.; Brewer, M. “Chlorodimethylsulfonium Chloride-Mediated Formation of Phenyl-a-chloroazoalkanes”, Synthetic Communications, 2008, 38, 3623-3630. (DOI: 10.1080/00397910802179961)
Javed, M.I., Brewer, M. “Diphenyldiazomethane”, Organic Syntheses, 2008, 85, 189-195. (DOI: 10.15227/orgsyn.085.0189)
Javed, M.I., Brewer, M. “Diazo Preparation via Dehydrogenation of Hydrazones with "Activated" DMSO” Organic Letters, 2007, 9, 1789-1792. (DOI: 10.1021/ol070515w)
Brewer, M. Conversion of hydrazones to alkyl chlorides under Swern oxidation conditions" Tetrahedron Letters, 2006, 47, 7731-7733. (DOI: 10.1016/j.tetlet.2006.08.120)
II. C-C Bond fragmentation as a route to medium sized rings and alkaloid natural products:
Several years ago we discovered a reaction that allows us to easily break a carbon-carbon bond while providing two new useful moieties (a carbon-carbon triple bond in conjugation with a carbonyl (i.e. an ynoate) and an aldehyde; 8 to 9, Scheme 4). We also showed that the new functionalities (aldehyde and ynoate) can be taken advantage of in further synthetic transformations including intramolecular 1,3-dipolar cycloadditions that give structurally-complex nitrogen-containing heterocycle products (e.g. 9 to 10, Scheme 4). Nitrogen-containing heterocycles are ubiquitous in biologically active compounds and their success as medicinal agents is indisputable. While there is a long history of research devoted to developing methods for the synthesis of nitrogen-containing heterocycles, structurally complex polycyclic heterocycles are still challenging to prepare and more efficient and concise methods for the preparation of these important compounds are needed.
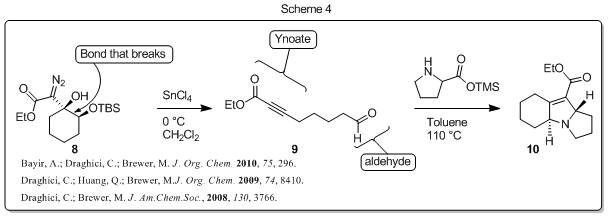
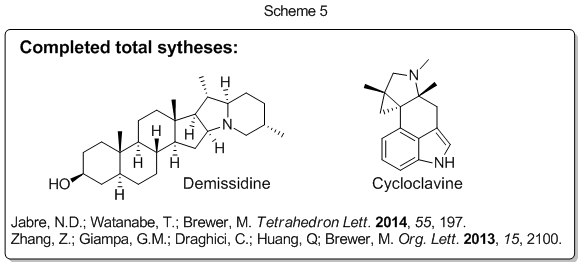
The synthetic strategy we developed (Scheme 4) allows us to prepare efficiently and rapidly complex molecular scaffolds that are present in many medicinally important compounds and natural products. We have shown that the scope of this reaction sequence is quite general and we have recently applied this methodology in the total synthesis of two unrelated alkaloids (demissidine and cycloclavine, Scheme 5). In addition, we have made significant progress toward the total synthesis of a third alkaloid natural product (aspidospermine; unpublished results).
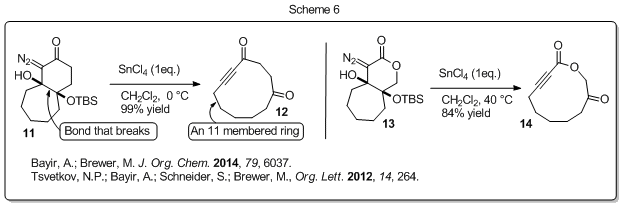
We have also successfully used this fragmentation reaction to prepare medium-sized-rings by engineering a system in which the ring fusion bond of bicyclic ring systems break over the course of the reaction. For example, bicycle 11 (Scheme 6) fragmented to provide the 10-membered ring 12 in 99% yield, and diazo lactone 13 fragmented to give ynolide 14 in 84% yield. We are currently applying this methodology to the synthesis of the natural product phoracantholide I.
Related Publications:
Bayir, A.; Brewer, M. “The fragmentation of bicyclic γ-silyloxy-β-hydroxy-α-diazolactones as an approach to ynolides” The Journal of Organic Chemistry, 2014, 79(13), 6037-6046. (DOI: 10.1021/jo500634d)
Jabre, N.D.; Watanabe, T.; Brewer, M. “Formal and Total Synthesis of (±)-Cycloclavine” Tetrahedron Letters, 2014, 55(1), 197-199. (DOI: 10.1016/j.tetlet.2013.10.152)
Zhang, Z.; Giampa, G.M.; Draghici, C.; Huang, Q; Brewer, M. “Synthesis of demissidine by a ring fragmentation/1,3-dipolar cycloaddition approach”, Organic Letters, 2013, 15(9), 2100-2103. (DOI: 10.1021/ol4004993)
Jabre, N.D.; Brewer, M.; “Stereoelectronic Effects in the Fragmentation of γ-Silyloxy-β-hydroxy-α-diazocarbonyl Compounds”, The Journal of Organic Chemistry 2012, 77(21), 9910-9914. (DOI: 10.1021/jo301944t)
Tsvetkov, N.P.; Bayir, A.; Schneider, S.; Brewer, M. “A Ring Fragmentation Approach to Medium-Sized Cyclic 2-Alkynones”, Organic Letters, 2012, 14(1), 264-267. (DOI: 10.1021/ol2030422)
Bayir, A.; Draghici, C.; Brewer, M. “Preparation of Tethered Aldehyde Ynoates and Ynones by Ring Fragmentation of Cyclic gamma-Oxy-beta-hydroxy-alpha-diazo Carbonyls”, The Journal of Organic Chemistry, 2010, 75 (2), 296-302. (DOI: 10.1021/jo902405f)
Draghici, C.; Huang, Q.; Brewer, M. “An Efficient Synthetic Approach to Polycyclic 2,5-Dihydropyrroles from α-Silyloxy Ketones”, The Journal of Organic Chemistry, 2009, 74 (21), 8410–8413. (DOI: 10.1021/jo901978y)
Draghici, C.; Brewer, M. “Lewis acid promoted carbon-carbon bond cleavage of d-silyloxy-β-hydroxy-α-diazoesters”, Journal of the American Chemical Society, 2008, 130(12), 3766-3767. (DOI: 10.1021/ja801004d)
III. Cross-College Collaboration for the Discovery of Novel Anti-anxiety Chemotherapeutics
Animal studies have shown that pituitary adenylate cyclase activating polypeptide (PACAP) and its receptor (PAC1) are involved in stress and anxiety related disorders. It has also been shown that PACAP blood levels are elevated in female patients who suffer from PTSD. Importantly, a truncated form of PACAP (PACAP6-38), a known antagonist of PAC1, blunts stress- and pain-induced anxiety in animal models. Unfortunately, PACAP6-38 is not a viable drug candidate. The goal of this cross-college collaboration is to discover small molecule antagonists of the PAC1 receptor as therapeutics for anxiety related disorders including PTSD.
This collaboration involves three research groups: Prof. Victor May's (Dept. of Neurological Sciences), Prof. Jianing Li's (Department of Chemistry), and mine. Prof. May is an expert in the field of cellular and molecular functions of PACAP signaling and the behavioral roles of PACAP in stress-mediated anxiety-related disorders, and Prof. Li is an expert in computational protein modeling with experience in modeling protein binding sites. Prof. Li has recently identified two potential binding sites within the PAC1 receptor. With this knowledge in hand, my group has begun synthesizing a series of compounds that will be tested by Prof. Victor May for their ability to act as PAC1 receptor antagonists. Through iterative cycles of design, synthesis and testing we hope to ultimately develop therapeutic strategies for stress and anxiety management.