
RPO | Human Subjects Research | IRB Policies and Procedures
Search for a Word
To search for a word use the keyboard shortcuts CTRL-F or F3. Mac users should use Apple's Command key (⌘) + F.
Table of Contents
- 1. Committee Mission
- 1.1 Introduction to the Boards, the Principles, and their Authority
- 1.2 Committee Membership (Sec. __.107)
- 1.3 Operations of the IRB (Sec. __.108)
- 1.3.1 Medical Safety Subcommittee Review
- 1.3.2 Guidelines for Continuing Review of Research
- 1.3.3 Guidelines for Review of Modifications
- 1.3.4 Conducting IRB Business in the Event of a Pandemic or Other Significant Emergency
- 1.3.5 IRB Minutes
- 1.4 Public Records and Open Meetings (Vermont Law)
- 1.5 IRB Jurisdiction
- 2. Institutional Ancillary Reviews
- 3. IRB Review Categories (Sec._46.109(a))
- 3.1 Full Committee Review
- 3.2 Expedited Review (Sec._46.110)
- 3.3 Limited Review (Sec._46.104)
- 3.4 Exemption Determination
- 3.5 Not Human Subjects Research Determination
- 3.6 Projects Not Requiring IRB Review
- 3.6.1 Case Studies
- 3.7 Determination of Institutional Engagement in Research
- 3.8 De Novo Review of Protocols
- 4. IRB Review Determinations 46.109 and 46.113
- 4.1 Administrative Hold, Suspension, or Termination of IRB-Approved Approvals
- 5. Eligibility to Perform Research At UVM/UVMMC
- 5.1 Responsibilities of Principal Investigators
- 5.2 Key Personnel Responsibilities
- 5.3 Access To and Retention of Research Records
- 5.4 CITI Training Requirements
- 5.5 Guidance on Data Management in Human Subjects Research
- 5.6 Managing Research Prior to Departure, Sabbatical, Medical Leave, or Other Absence
- 5.7 Data and Biospecimen Sharing
- 5.8 Enrollment Incentives
- 6. Conflict of Interest
- 7. Non-Faculty Researcher Requirement
- 7.1 Student Class Project Guidelines
- 8. Types of Research
- 8.1 Standard Clinical Trial Protocol
- 8.1.1 Elements Found in a Standard Protocol
- 8.1.2 Requirement to include a full Protocol Title in the Electronic Medical Record (EPIC)
- 8.1.3 Plans for Recruitment/Screening/Retention
- 8.1.4 Participant Compensation
- 8.2 Chart Review Protocol
- 8.3 Qualitative Research Protocol
- 8.4 Biological Specimens/Data Repository Protocols
- 8.4.1 Research Tissue Acquisition Policy
- 8.5 Blood Collection Protocols 46.110
- 8.5.1 Blood Drawing Limits
- 8.6 Research Involving Coded Private Information or Biological Specimens
- 8.7 Sustainable Agriculture Research and Education (SARE) Grant Projects
- 8.8 Department of Defense (DoD) Supported Research Projects
- 8.9 Exception from Informed Consent Emergency Research
- 8.9.1 Review Flow for Planned Emergency Research
- 8.9.2 Designing a Plan for Community Consultation and Public Disclosure in Planned Emergency Research
- 8.9.3 Data Collection Expectations for Exception from Informed Consent for Emergency Research
- 8.9.4 Operations of Exception from Informed Consent for Emergency Research Advisory Panel
- 8.10 Research Conducted in Public Schools
- 8.11 International Research: Information on Conducting Research Outside of the United States
- 9. Consent (§____.116)
- 9.1 Children
- 9.1.1 Children Reaching Legal Age of Consent While Enrolled in a Study Policy
- 9.2 Surrogate Consent for Research (Legally Authorized Representatives)
- 9.3 Cases of Physical Compromise
- 9.4 Informed Consent and HIPAA Authorization Process for Non-English Speaking Individuals
- 9.5 Waiver of Informed Consent, Alteration of Informed Consent, or Waiver of Documentation §____.116(c)
- 9.6 Consent Process for Legally Blind or Impaired Vision Research Participants
- 9.7 Guidance for Researchers Using Deception or Incomplete Disclosure in Research
- 9.7.1 Debriefing
- 9.7.2 Additional IRB Considerations when using Deception or Incomplete Disclosure in Research
- 9.8 Media Consent
- 9.9 Obtaining Electronic Written Consent
- 9.10 Telemedicine and Research Visits
- 9.11 Technical Guidance for Remote Visit
- 10. Data Protection Regulations
- 10.1 Health Insurance and Portability Act (HIPAA)
- 10.2 Waiver, Partial Waiver or Alteration of HIPAA Authorization 45 CFR 164.512
- 10.3 European Union (EU) Participants and EU General Data Protections (GDPR)
- 10.4 Decedent Data
- 10.5 National Institutes of Health Genomic Data Sharing Policy
- 11. Certificates of Confidentiality
- 12. Exceptions to Confidentiality Guidance
- 13. Cooperative Research (Single IRB) Policy (Sec. __.114)
- 13.1 Cooperative Research (Single IRB) Roles and Responsibilities
- 13.2 Collaborative Agreements
- 13.3 Procedures for Reliance on External IRB (Sec __.114(b)(1))
- 13.4 Non-Collaborative Review and UVM IRB
- 13.5 Procedures for Reviewing or Relying for NNE-CTR Grants (Sec __.114(b)(1))
- 13.6 Collaborative Research Between UVM and the VT Agency of Human Services (AHS)
- 13.7 Collaborations with Community Partners
- 14. Funding/Contracts/Fees
- 14.1 When the Project is a New Competing or a Competing Renewal Application and the New Protocol is Identical or Substantially Similar to an Approved Protocol
- 14.2 IRB Review of Just-in-Time (JIT) Protocols
- 14.3 Grant Proposals Lacking Definite Plans for Involvement of Human Subjects
- 14.4 Contracts/Agreements
- 14.5 Changes to the Scope of a NIH Awarded Project
- 14.6 Fees for Committee on Human Research Review of Sponsored Trials
- 15. Modification to Previously Approved Protocol
- 16. Continuing Review Requirements
- 17. Closing a Protocol by the PI
- 17.1 Closure of Protocol by Committee
- 18. Reportable New Information (includes unanticipated problems and adverse events)
- 18.1 Reportable New Information on External IRB Studies
- 19. Incidental Findings in Neuroimaging Protocols – Detection and Management
- 19.1 Standards and Language for Studies Involving MRI
- 20. Investigational Drugs (including Biologics)
- 20.1 Use of Approved Drugs for Off-Label Indications
- 20.2 Expanded Access of Investigational Drugs (Compassionate Use)
- 20.3 Food and Food-Derived Products, Spices/Herbs, or Dietary Supplements
- 20.4 Controlled Substances Used in Research
- 21. Investigational Devices
- 21.1 Expanded Access of Investigational Devices
- 22. Humanitarian Use Device (HUD) Designation and Humanitarian Device Exemptions (HDE)
- 23. Emergency Use of an Investigational Drug or Biologic or Investigation Device
- 24. Subjects Vulnerable to Coercion or Undue Influence
- 24.1 Pregnant Women, Fetuses, Neonates of Uncertain Viability and Non-Viable Neonates 45 CFR 46 Subpart B
- 24.2 Research Involving Prisoners - Subpart C
- 24.3 Children
- 24.4 Non-English Speaking Individuals Participating in Research
- 25. Pregnancy Testing in Minor Research Subjects
- 26. Human Subject Quality Assurance Reviews
- 26.1 Human Subject Quality Assurance Reviews on External IRB Studies
- 27. Noncompliance Policy and Procedures
- 27.1 Non-Compliance Review Procedures
- 27.2 Definitions, Guidance, and Examples Referenced as Part of Non-Compliance Reviews
- 27.3 Non-compliance Policy on External IRB Studies
- 28. Electronic Signatures Policy
- 29. Regulatory Definitions
- 30. Statement of Compliance for the Committees on Human Research
- 31. 2018 Common Rule Transition
Section I Committee Information
1. Committee Mission
The University of Vermont and UVM Medical Center are responsible for safeguarding the rights and welfare of human subjects involved in any research activity. According to institutional policy, all such research, funded or unfunded, conducted by University and/or UVM Medical Center personnel, including students, or done under the auspices or sponsorship of either institution must be reviewed by one of the Institutional Review Boards (IRBs): the Committee on Human Research in the Medical Sciences (CHRMS) or the Committee on Human Research in the Behavioral and Social Sciences (CHRBSS). Approval must be obtained BEFORE the research activity starts and the project must be reviewed at least annually for as long as it is active.
1.1 Introduction to the Boards, the Principles, and their Authority
Revised 10.12.2023
The Institutional Review Boards (IRBs) at the University of Vermont (UVM) serve UVM and the University of Vermont Health Network (UVMHN). UVM and UVMHN each have their own Federalwide Assurances of Compliance with DHHS Regulations for the Protection of Human Subjects (FWA).
• The “Committee on Human Research in the Medical Sciences" (CHRMS) is authorized to review all proposals to use human subjects in biomedical research. CHRMS also functions as the Privacy Board for UVMHN for the Health Insurance Portability and Accountability Act (HIPAA) by reviewing all authorization, partial waivers or requests to waive written authorization for research undertaken at both institutions.
• The “Committee on Human Research in the Medical Sciences (CHRMS II), is a subset of the full committee (CHRMS) for situations where it is not practicable to convene the full CHRMS.
• The “Committee on Human Research in the Behavioral and Social Sciences" (CHRBSS) is authorized to review all proposals to use human subjects in the behavioral and social sciences. CHRBSS may also function as the Privacy Board for UVMHN for the Health Insurance Portability and Accountability Act (HIPAA) by reviewing all authorizations, partial waivers, or requests to waive written authorization for research undertaken at both institutions. CHRBSS also reviews Reportable New Information (RNI’s) such as, adverse events, protocol and consent deviations, unanticipated problems etc.
• The Safety Subcommittee, which is a subset of the full Committees, reviews Reportable New Information (RNI’s) such as, adverse events, protocol and consent deviations, unanticipated problems etc. Minutes from the meeting are distributed to the Full CHRMS IRB Committee each month.
• Ad-hoc Noncompliance Subcommittees, including a subset of the members and other institutional personnel as applicable, are convened as necessary to review noncompliance cases.
• The IRB Policy and Procedure Subcommittee, includes Committee leadership, IRB members and IRB regulatory analysts, convenes monthly to develop, revise and review IRB policies and human subject regulations. Notes from the meeting are distributed to the Full IRB CHRMS Committee each month.
• Exception from Informed Consent for Emergency Research Advisory Panel (Section 8.9.4)
• Additional subcommittees may be added as needed.
The Committees have been established to review all research projects and activities involving human subjects in accordance with the Federalwide Assurances that both UVM and UVMHN have in place to ensure the rights and welfare of those involved are adequately protected, that the methods used to obtain informed consent are adequate and appropriate, and any risks to research participants are out-weighed by the potential benefit to them or by the general importance of the knowledge to be gained.
Governing Principles
The Committees are governed by the basic principles regarding experimentation on humans which have their origins in the Nuremberg Code, the Belmont Report, and the Declaration of Helsinki and are consistent with the regulations governing research with human subjects, i.e., 45 CFR 46 of the Code of Federal Regulations.
The institutions conduct clinical research that comes under the jurisdiction of the US Food and Drug Administration (FDA). When conducting this research, UVM and UVMHN comply with the applicable sections of the CFR (usually 21 CFR 50 and 21 CFR 56).
The institutions conduct research funded by the Department of Defense (DoD). The DoD regulations for the protection of human subjects are applied when conducting, reviewing, approving, overseeing, supporting, or managing DoD-supported research with human subjects. See section 8.8 of the manual for information about how UVM and UVMHN applies the DoD regulations.
The IRB follows the Health Insurance Portability and Accountability Act (HIPAA) provisions (45 CFR 160, 162, & 164) for the protection of individually identifiable health information used in research. See section 10 of the manual for information how the regulations are applied.
Federalwide Assurance
UVM and UVMHN each have established Federalwide Assurances with the Department of Health and Human Services (DHHS) that commits the institutions to comply with the requirements in the HHS Protection of Human Subjects regulations at 45 CFR part 46.
The FWA application includes the option to apply all the regulations at 45 CFR 46 and all the Office for Human Research Protections (OHRP) requirements regardless of the funding source for the research. UVM and UVMHN each have chosen to “uncheck” the box which means the FWAs for UVM and UVMHN are limited in applicability to federally sponsored or conducted research. This enables the IRB to exercise additional flexibility. For instance, for non-federally supported research, reporting of suspensions, terminations, unanticipated problems involving risk to subjects or others, and serious and continuing noncompliance to OHRP and federal agencies and departments as mandated by .108(a)(4)(i-ii) and .113 is not required.
In general, UVM and UVMHN do extend the principles of the regulations governing research with human subjects to research not federally sponsored or conducted. The IRB reserves the right to review any other research involving human subjects not otherwise covered by the FWA that is conducted at UVM or UVMHN facilities.
Prompt reporting for all research conducted or supported by any federal department or agency will take place in accordance with the regulations governing research with human subjects, i.e., 45 CFR 46 of the Code of Federal Regulations and for FDA regulated clinical trials, the applicable sections of the CFR 21 CFR 50 and 21 CFR 56.
As per the 2018 Common Rule change, the University is not required to designate one or more IRBs on its FWA Sec.__.103(b)(2).
Committee Authority
The President of the University (UVM) has delegated the authority to the Vice President for Research as the lead Institutional Official (IO) responsible for the assurance of compliance in the area of human subject protections. The Committees are established by authority of the Vice President for Research. The Committees are delegated the specific authority to:
• (UVM)Define the basic policies, procedures and standards by which human research protocols will be reviewed;
• Sec. __109(a)Review and have authority to approve, require modifications in, or disapprove all research activities, including exempt research activities under Sec. __.104 for which limited IRB review is a condition of exemption
• Sec. __109 (b)Require that information given to subjects (or legally authorized representatives, when appropriate) as part of informed consent is in accordance with Sec. __.116. The IRB may require that information, in addition to that specifically mentioned in the regulations, be given to the subject when, in the IRB's judgement, the information would meaningfully add to the protection of the rights and welfare of subjects.
• Sec. __109 (c) Require documentation of informed consent or waive documentation in accordance with the regulations.
• Sec. __109 (e) Conduct continuing review of research at intervals appropriate to the degree of risk, but not less than once per year, and shall have authority to observe or have a third party observe the consent process and research.
• Sec. __108 (4i)Investigate and report to the appropriate institutional officials, Office of Human Research, DHHS (OHRP), and, when applicable US Food and Drug Administration (FDA) and/or funding agency, any serious or continuing noncompliance with the federal regulations and requirements and determinations of the IRB.
• Sec. __113 Suspend or terminate approval of research that is not being conducted in accordance with the IRB's requirements or that has been associated with unexpected serious harm to subjects. Any suspension or termination of approval shall be reported promptly to the investigator, appropriate institutional officials, OHRP, and/or FDA when applicable.
• (UVM)Provide training for all individuals involved in the conduct of research involving human subjects, regardless of funding source.
• (UVM)Review all adverse events and unanticipated problems to subjects and others meeting local IRB criteria.
• (UVM and UVMHN) Function as the Privacy Board by reviewing all HIPAA authorization language or requests to waive authorization for research undertaken at both UVM and UVMHN.
• Sec. __111 Monitor active protocols to ensure compliance with the IRB-approved protocol and with applicable human subject protection guidelines and regulations.
The authority of the Committees to disapprove, restrict, suspend or terminate a human research study may not be overridden. However, the Vice President for Research will have the final authority to disapprove, restrict, suspend or terminate a study, which has received Committee approval.
Resources
(Sec. __108(a)(1)The institution will provide IRBs with resources, office space, professional staff, and support staff sufficient to carry out their responsibilities efficiently and effectively and to serve as day-to-day liaison with appropriate University administrative offices, project investigators, other institutional safety and ethics boards, and various regulatory and funding agencies.
Institutional Relationships
UVM is the institution associated with the IRB registrations for the Committees with the Office for Human Research Protections (OHRP) and has a written agreement with UVMHN for their reliance on UVM’s IRBs. The Research Protections Office (RPO) is responsible for completing the IRB registration in accordance with 21 CFR 56.106 and 45 CFR 46 subpart E. The Vice President for Research is the designated Institutional Official for research involving human subjects for UVM. The President and CEO of the University of Vermont Health Network is the Institutional Official. The Committees report to regulatory authorities (i.e., FDA, OHRP) through the institutional official(s) or his/her designee at UVM and/or UVMHN, as appropriate.
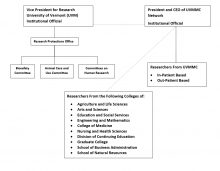
1.2 Committee Membership (Sec. __.107)
Revised 6/1/2023
The membership of the Committees meets all criteria required by the applicable regulations and guidelines governing research with human subjects. The Committees are constituted of members with varying backgrounds sufficiently qualified for review through appropriate experience and expertise. When membership decisions are made, consideration is given to gender, race, cultural backgrounds, and sensitivity to community attitudes.
Committee leadership and members serve at the discretion of the Institutional Official (IO). The IO has delegated signature authority to the Executive Director of Research for the appointment letters.
Chair
- Committee Chairs are appointed by the IO.
- A Committee Chair must be a current or Emeritus University faculty member and must have prior service as a Committee member.
- It is the responsibility of the Committee Chairs to conduct Committee meetings in accordance with established federal regulations and University operating policies and procedures. These responsibilities, which include but are not limited to the following, are to:
- sign official Committee action documents and human subjects assurance forms;
- keep abreast of relevant state and federal regulations;
- meet as needed with the IO to discuss Committee activities;
- meet as needed with each other to coordinate the efforts of the Committees;
- keep abreast of procedures for maintenance of official protocol files and other administrative operations of their respective Committees;
- recommend, in consultation with the Director of the IRB, new members to the IO;
- ensure that new members are properly oriented to and educated about their duties and responsibilities;
- initiate activities designed to assist investigators and keep the campus and community apprised of their rights and responsibilities with regard to human research; and
- assist appropriate University administrators in the preparation of federal reports and assurances and meet with federal IRB auditors as necessary.
- No method for removal is delineated, as all members are appointed and serve at the discretion of the IO.
- Appointment to the Chair will be for renewable two-year terms.
Associate Chair
- The Associate Chair must be appointed from the regular voting membership
- The Associate Chair will conduct the meetings of the IRB in the absence of the Chair or if there is a conflict by the Chair’s participation
- The Associate Chair will assist in the conduct of expedited protocol reviews
- Associate Chair will sign official Committee action documents and IRB approvals in the Chair’s absence
- The Associate Chair is appointed for renewable two-year terms.
- The Associate Chair of CHRMS will be one of the licensed physicians on the Committee if the Chair is not a licensed physician.
Regular Members
Members will be of varying professional and personal backgrounds and must demonstrate a genuine interest in and commitment to the purpose of the Committees. Specific membership criteria will comply with all relevant federal and state regulations. Every effort will be made to fulfill principles which embrace cultural diversity. The Committee Members’ duties are delineated in subsequent sections.
- The IO appoints all Committee Members, after receiving recommendations from the IRB Chair and the IRB Director. When vacancies occur, nomination will be sought, after which formal recommendation(s) for new member(s) will be made by the Committee Chair to the IO.
- The IO will in no case make a final appointment without prior consultation with the Committee Chair and/or the IRB Director.
- Committee appointments will usually be for renewable two-year terms, serving at the discretion of the IO.
- Consideration is given to achieve a balance between new and experienced members when determining which appointments will be renewed.
- No specific attendance requirements are delineated; however, it is required that committee members demonstrate a genuine interest and commitment to the purpose of the Committees.
- No method for removal is delineated, as all members are appointed and serve at the discretion of the IO.
- (Sec.__107)(b) Each IRB shall include at least one member whose primary concerns are in scientific areas and at least one member whose primary concerns are in nonscientific areas.
- (Sec.__107(c) Each IRB shall include at least one member who is not otherwise affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution.
- (Sec. __107(d) No IRB may have a member participate in the IRB's initial or continuing review of any project in which the member has a conflicting interest, except to provide information requested by the IRB.
- (Sec. __107(e) An IRB may, in its discretion, invite individuals with competence in special areas to assist in the review of issues that require expertise beyond or in addition to that available on the IRB. These individuals may not vote with the IRB.
Ex Officio Members
An ex officio member is defined as a member who serves by virtue of an office or position held outside the IRB Office. An ex officio member may be appointed by the IO as a voting member, a non-voting member, or an alternate member.
- University of Vermont Medical Center HIPAA Privacy and Security Specialist will serve ex officio as a regular member with full voting privileges.
- The University of Vermont Chief Information Officer will serve ex officio as a regular member with full voting privileges.
- The RPO Director will serve as an ex officio member. This individual may serve as an alternate member if so appointed.
- Additional ex officio members may be appointed at the discretion of the IO.
Alternate Members
An alternate member is defined as a member who substitutes for a specific member or members with similar qualifications, experience, or membership category. When an alternate member substitutes at a meeting, they appear as “substitutions” on the minutes. Alternate members may be appointed under the following conditions:
- Must be appointed by the IO and listed in IRB rosters submitted with regulatory documents.
- Must be designated to serve as the alternate for a specific member or members who have the same attributes (e.g., scientific member can only substitute for another scientific member).
- Alternates must receive same onboarding training as non-alternate members.
- Must receive all proposal materials in advance of the meeting for review if they will be voting during the meeting.
- Alternate members are advised to "vote their conscience" as opposed to representing the position of the regular member for whom they serve.
- If both the regular voting and alternate member both attend a meeting, only the regular voting member may vote. An alternate member may only be required to vote when necessary to achieve or maintain quorum.
- The IRB Director IRB Regulatory Analysts, and IRB Reliance Administrator are alternate members. They are designated by the chair to review and approve minor changes to research, continuing reviews of minimal risk research, make exempt determinations and grant waivers and alterations of HIPAA.
Regular Voting Membership of the Committee on Human Research in the Medical Sciences (CHRMS)
It is recommended that CHRMS be composed minimally of 12 regular members. This may include ex officio members who have regular appointments with full voting privileges.
- Of the total, it is recommended that 8 regular members be representatives of appropriate scientific, academic, and clinical research disciplines, and which may include but are not limited to the following:
- a pharmacist or pharmacologist
- a psychiatrist
- a nurse, allied health professional, or nutritionist
- a researcher holding a Ph.D. degree in a basic biomedical science
- a pathologist or a pathology researcher associated with the UVM/UVMMC pathology services
- two licensed physicians from other appropriate clinical disciplines (such as pediatrics, surgery, oncology, orthopedics, neurology, obstetrics, and gynecology)
- Of the total, it is recommended that 4 regular members be non-scientific representatives. Non-scientists may include but are not limited to the following:
- individuals whose backgrounds and perspectives will help ensure a well- rounded and objective review board, e.g., community attorney, ethicist
- Of the total, the membership must include at least one individual with no formal affiliation with UVM or UVMMC.
CHRMS II Membership
This Committee consists of five members, with all other CHRMS I members designated as alternates. The Chair of this committee will be the same person as that designated by the CHRMS I committee.
Regular Voting Membership of the Committee on Human Research in the Behavioral Sciences (CHRBSS)
It is recommended CHRBSS be composed minimally of 8 voting members, 2 to be nonscientific representatives, and the remainder to be representatives of appropriate scientific, academic, or clinical disciplines. This may include ex officio members who have regular appointments with full voting privileges.
The scientific/academic/clinical representatives should include persons involved in research in the appropriate disciplines, which may include but is not limited to:
- psychology
- sociology/anthropology
- education/social services
- appropriate biomedical fields
- other social sciences
Of the total, the membership must include at least one individual with no formal affiliation with UVM.
Consultation
The Committees may, at their discretion, obtain consultation from individuals with expertise in specialized areas to assist in the review of complex issues which require expertise beyond or in addition to that available on the IRB. The IRB shall consult with General Counsel and other University Officials or Hospital Officials, as indicated, to address issues pertaining to institutional policies, applicable law, and standards of conduct and practice. These individuals are not allowed to vote.
General liability insurance coverage
Actions by members carried out as a function of their committee appointments are included under the University's general liability insurance coverage.
Monetary compensation
Appointment to the Committees is without monetary compensation.
Conflict of Interest
See Section 6 Conflict of Interest requirements and management of conflicts for committee members.
Confidentiality and Code of Conduct
This Code of Conduct is a set of behavioral expectations intended to assure that our Committee members uphold the highest level of integrity and ethical standards. Members must not discuss, disclose, or reproduce any protocol-related information, except as necessary to carry out responsibilities or as required by law. Members must limit their electronic access to that which is required to fulfill their Committee duties. Members must never access any research protocols to satisfy personal interest or curiosity. Any printed materials for review should be returned to the IRB office or shredded after use.
IRB Member Training
Before a new Committee member can be added to the roster and vote at a convened meeting, they must complete the following items:
- An initial orientation meeting is held with the Chair and/or IRB Director. The initial orientation meeting includes review of human subject protection documentation including the IRB, Policy and Procedures, Reviewer Materials, the electronic submissions and reviewer software, and the RPO Website.
- New Committee members are educated on the IRB code of conduct, expectations for written protocol reviews, and maintaining quorum.
- Federal and local regulations along with institutional policy governing human subject research is also reviewed.
- Examples of pre-review materials and current journal articles on research may be given as additional educational material as well.
- Completion of the Human Subjects in Research Training Module through CITI. Prison representatives are not required to complete the CITI training.
Before a new member can be assigned to review a protocol, they must complete their mentorship as described below. Prior experience on an IRB may substitute for some or all of the mentorship program requirements, as determined by the IRB Chair.
Member Mentorship Program
First Meeting: New member observes the meeting and may vote.
Second Meeting: New member is assigned a “Shadow” review and receives the same materials as the assigned reviewer. The new member does not need to present anything to the committee but can compare their review with the experienced reviewers as part of the learning process and may vote.
Third Meeting: New member is assigned a review and is paired with an experienced reviewer (primary reviewer) to do a “shadow” review. The new member should consult with the experienced reviewer, the staff, or the Chair if there are any questions or concerns during the review. The new member will present to the committee as the third reviewer and may vote. The experienced reviewer should be prepared to “jump in” and provide assistance if there are any concerns during the review presentation at the meeting.
Fourth Meeting: New Member completes an independent review and presentation to the Full Committee.
Member mentorship may take more meetings than the outlined timeline and certain steps may be repeated as needed, as mentorship is dependent upon the number of submissions discussed at committee and reviewer availability.
Documentation of Training Completion
Records of completion dates are maintained in RPO shared folder S:\irb\Committee Member Management\Rosters.
Continuing Education
Continuing education is accomplished by retaking the Human Subjects in Research tutorial at least once every three years, attendance at webinars, regional or national meetings and conferences. Additional education is provided as topics discussed during the monthly Committee meetings.
1.3 Operations of the IRB (Sec. __.108)
Revised 12/02/22
Convened Meetings
The CHRMS and CHRBSS Committees meet monthly when there are agenda items.
CHRMS II is only convened when it is impracticable to convene a full meeting. This decision will be made by the IRB Regulatory Analysts in consultation with the Committee Chair. CHRMS II will not be used to conduct regular routine business and will follow all policies, procedures, and guidelines of the full committee.
In-Person or Virtual Convened Meetings
Convened meetings may occur all in-person, all virtually or a mix of both. Offering virtual and hybrid meetings provides easier access to the meeting for many members. Use of an electronic submission and review process readily supports the virtual review process. The regulatory requirements (e.g., quorum, representation, etc.) for both types of meetings are met and documented in the minutes.
The IRB will either schedule a suitable conference room or utilize Microsoft Teams for the IRB meetings. Meeting proceedings are conducted in the same order/manner in both types of meetings. The agenda items are discussed, a motion is made, and members cast their audible votes.
Members who are conflicted with a specific agenda item must recuse from discussion and cannot count toward a quorum or vote on that item per Section 6. Conflict of Interest. Members should notify IRB staff of their conflict prior to the meeting, when possible, so IRB staff can determine if quorum can be maintained during the recusal on that specific vote. Both in-person and virtual meetings require the conflicted member to step out (physically or virtually) temporarily during the discussion and vote on the given agenda item. They are admitted back into the meeting once the vote is complete.
Meeting Notices
The agenda, including the time and location of the meeting, are distributed in advance to all members. All pre-meeting materials for protocols are located within the electronic system.
Non-Member Attendance at a Convened Meeting
Guests are allowed to attend a convened meeting in certain circumstances (e.g., students for educational purposes or consultants for expertise opinion). These individuals are required to complete the IRB Meeting Guest Confidentiality Agreement prior to attending a meeting. Principal investigators are also allowed and sometimes invited to a convened meeting to address/discuss human subject issues with their proposed or ongoing protocol.
Conducting Initial Reviews
Initial convened IRB reviews and approvals will occur in compliance with 45 CFR 46, 21 CFR 50 AND 56 when applicable, and provisions of this Assurance for each project not properly found to be exempt (Section 101[b]) by the Research Protections Office.
The Committees determine all of the requirements outlined in 45 CFR 46.111 are satisfied for each protocol, including all of the requirements for obtaining informed consent and documentation of written informed consent as applicable. Reviewer forms and checklists are utilized as a guide by reviewers and other Committee members to ensure that these criteria have been met.
The IRB shall apply additional protections as necessary to protect potentially vulnerable research subjects. Not every human being is capable of self-determination. The capacity for self-determination matures during an individual's life, and some individuals lose this capacity wholly or in part because of illness, impaired decision-making capabilities, or circumstances that severely restrict liberty. The extent of additional protection afforded depends upon the risk of harm and the likelihood of benefit. In addition, when IRB reviews research involving a specific vulnerable population, consideration will be given to inclusion of one or more members who are knowledgeable about and experienced in working with these subjects. All specific IRB findings as required by 45 CFR 46 for special protections will be documented.
IRB and federal regulation policy require a prisoner representative to be present and review the protocol at a meeting when a prisoner population is the target of the research.
All initial reviews conducted at a convened meeting will have assigned a primary and a secondary member reviewer. Exceptions to this will be if the committee reviews a HUD or an Expanded access protocol in which only one primary reviewer is required.
Conducting Continuing Reviews
Continuing convened IRB reviews and approvals will occur in compliance with 45 CFR 46, 21 CFR 50 AND 56 when applicable, and provisions of this Assurance for each project not properly found to be exempt (Section 101[b]) by the Research Protections Office. Continuing reviews, as applicable, will be preceded by IRB receipt of appropriate progress reports from the investigator, which incorporate relevant study-wide findings.
The Committees determine all of the requirements outlined in 45 CFR 46.111 continue to be satisfied for each annual continuing review submission, including all of the requirements for obtaining informed consent and documentation of written informed consent as applicable. Reviewer forms and checklists are utilized as a guide by reviewers and other Committee members to ensure that these criteria have been met.
Continuing reviews may be approved pending requests for minor protocol edits or clarifications but are not released to the researcher until receipt of an acceptable response to the clarifications. In some instances, this process results in a continued approval date that is prior to the response date. All reviews are conducted electronically through the electronic submission and review system.
All continuing reviews conducted at a convened meeting will have assigned one primary member reviewer.
Written Review Procedures
The institution and the designated IRBs have established written procedures for the following, in accordance with the terms stated in the FWA. (Sec. __.108)(3)
· verifying whether proposed activities qualify for exemption from IRB review;
· conducting IRB initial and continuing review, approving research, and reporting IRB findings to the investigator and the institution in writing as required;
· determining which projects require review more often than annually, and which projects need verification from sources other than the investigator that no material changes have occurred;
· ensuring that changes in approved research are reported promptly and are not initiated without IRB approval, except when necessary to eliminate apparent immediate hazards to the participant;
· ensuring prompt reporting to the IRB, institutional officials, the relevant Department or Agency Head, any applicable regulatory body, and OHRP of any
(i) unanticipated problems involving risks to subjects or others in any covered research;
(ii) serious or continuing noncompliance with Federal, institutional, or IRB requirements; and
(iii) suspension or termination of IRB approval for Federally supported research;
· official action on protocols involving use of drugs or medical devices is taken in accordance with applicable FDA regulations governing human research review; and
· the IRB has a process for monitoring on-going research to assess congruence with the IRB-approved protocols and compliance with applicable human subject protection guidelines and regulations, which includes monitoring a sample of studies, prioritizing the review of protocols that do not have other formal monitoring processes in place.
· Except when certain exempt or expedited review procedure is used (as described in Sec. __.110), an IRB must review proposed research at convened meetings at which a majority of the members of the IRB are present, including at least one member whose primary concerns are in nonscientific areas. In order for the research to be approved, it shall receive the approval of a majority of those members present at the meeting.
Approval, Effective, and Expiration Dates
Definitions:
Approval Date: The date on which the reviewer approved the study, continuing review, or modification as submitted without any conditions, or approved the study or modification conditionally, pending modifications. No unapproved research activities involving human subjects may be initiated until the approval becomes effective.
Effective Date: Whenever the IRB approves a research study, continuing review or modification with one or more conditions at the time of review, the effective date of the approval is the date on which the IRB chair or his/her designee has reviewed and accepted as satisfactory any revised protocol or informed consent documents, or any other responsive materials required by the IRB from the investigator. In these circumstances, no unapproved research activities involving human subjects may be initiated until the conditions have been satisfied in the manner set forth by the IRB and the approval becomes effective.
The IRB determines the initial approval and effective dates in the following manner:
Protocols reviewed at a Full convened meeting:
- When a research study is approved without any clarifications or revisions at a convened meeting, the date of the convened meeting is both the approval and effective date:
- Approval Date: the date of the committee meeting
- Effective Date: the date of the committee meeting
- Expiration Date: one year minus one day after the approval date. For logistical reasons, the IRB may set the expiration date less than 12 months.
- When the research study is approved but requires a response secondary to clarifications or revisions at a convened meeting:
- Approval Date: the date of the committee meeting
- Effective Date: the date the committee approved the response
- Expiration Date: one year minus one day after the approval date. For logistical reasons, the IRB may set the expiration date less than 12 months.
Protocols reviewed through the expedited review process:
- When a research study is reviewed by the Chair or his/her designee and approves the project without requiring any clarifications or revisions:
- Approval Date: the date that the reviewer made their initial determination.
- Effective Date: the date that the reviewer made their initial determination (note that in this case, since there are no modifications required, the approval and effective date will be the same.)
- Expiration Date: one year minus one day after the approval date.
- When a research study is reviewed by the Chair or his/her designee and requires clarifications or revisions to the protocol as a condition of approval:
- Approval Date: the date that the reviewer made their initial determination.
- Effective Date: the date that the Chair or his/her designee approved the response. (Note that in this case, since modifications were required, the effective date will not be the same as the approval date.)
- Expiration Date: one year minus one day after the approval date.
Modifications reviewed at a Full convened meeting:
- When a modification is approved without any clarifications or revisions at a convened meeting, the date of the convened meeting is both the approval and effective date:
- Approval Date: the date of the committee meeting
- Effective Date: the date of the committee meeting
- When the modification is approved but requires a response secondary to clarifications or revisions at a convened meeting:
- Approval Date: the date of the committee meeting
- Effective Date: the date the committee approved the response
Modifications reviewed through the expedited review process (including study team member updates):
- When a modification is reviewed by the Chair or his/her designee and approves the project without requiring any clarifications or revisions:
- Approval Date: the date that the reviewer made their initial determination.
- Effective Date: the date that the reviewer made their initial determination (note that in this case, since there are no modifications required, the approval and effective date will be the same.)
- When a modification is reviewed by the Chair or his/her designee and requires clarifications or revisions to the protocol as a condition of approval:
- Approval Date: the date that the reviewer made their initial determination.
- Effective Date: the date that the Chair or his/her designee approved the response. (Note that in this case, since modifications were required, the effective date will not be the same as the approval date.)
*Please note that the overall the effective date of a protocol (i.e. the effective date displayed on the parent study submission) is the effective date of the most current version of the protocol. This date will change each time a continuing review or modification has been approved.
Determining date for second and all subsequent continuing reviews
Continuing reviews reviewed at a Full convened meeting
For all subsequent continuing reviews of a research study requiring ongoing approval, the date of the convened meeting at which the IRB conducts continuing review and approves the study (with or without conditions) is the next approval date. The expiration date will be one year minus one day after the approval date. For logistical reasons, the IRB may set the expiration date less than 12 months.
The overall protocol effective date will be updated to reflect the effective date of the continued approval.
Continuing reviews reviewed through expedited review process:
For all subsequent continuing reviews of a research study requiring ongoing approval, the date that the Chair or his/her designee conducts continuing review and approves the study (with or without conditions) is the next approval date.
The expiration date will be one year minus one day after the approval date. The overall protocol effective date will be updated to reflect the effective date of the continued approval.
Voting Requirements
1. A majority of the total number of regular voting members will constitute a quorum. The number in attendance must be one more than half the total number of regular voting members. If less than a majority of the total number of regular voting members is present, one or more alternate members may be included to constitute a quorum if they have been specifically designated to alternate for a specific absent member and meet all other membership criteria.
2. At least one nonscientific member must be present to constitute a quorum.
3. For review of FDA regulated articles, at least one physician must be present to constitute a quorum.
4. Official Committee action on protocols involving human subjects will be by formal vote of a simple majority at convened meetings of a quorum of Committee members.
5. All meetings will be conducted using Robert’s Rules of Order as guidance, with deviations made as deemed appropriate by the Chair.
6. Official action on a protocol involving use of drugs or medical devices will be in accordance with FDA regulations governing human research review.
7. IRB members may participate in a convened meeting of the IRB via telephone or video conferencing. Those members have access to the research protocol materials in advance of the meeting within UVMClick-IRB.
Report Findings
The Committees on Human Research are responsible for reporting findings, actions as well as requesting clarifications to the investigators in writing, and to the appropriate offices within the institutions’ administration through reports and meeting minutes to institutional officials through their representatives on the Committee, and to sponsors of research, if so required.
Process for Appeal
There is no process delineated for appeal of Committee decisions. However, there is no prohibition for resubmission of specific requests or protocols for additional review by the Committee.
Protocols Requiring More Frequent Review
Determination of which studies require review more often than annually is done at the time of initial protocol review, continuing review, on a case-by-case basis, depending upon protocol specific factors, including, but not limited to, the level of risk.
Protocols Requiring Verification from Other Sources
Determination of which studies need verification from sources other than the investigators that no material changes have occurred since previous IRB review is done on a case by case basis either by the primary reviewer at initial review, continuing review, or through information received (e.g., adverse event or unanticipated problems to subjects or others reports or complaints) and would depend upon protocol specific factors. Information is also collected through the Committee’s monitoring program and through reports from both internal sources (i.e., Protocol Review and Monitoring Committee at the Vermont Cancer Center or the Clinical Research Center’s Scientific Advisory Committee or the Research Subject Advocate) and external entities (i.e., DSMBs or sponsor monitoring visit reports).
Modification to Protocol
The Committees on Human Research require changes in approved research to be reviewed and approved prior to initiation except where it is necessary to eliminate immediate hazard. Changes implemented to the protocol prior to Committee approval is considered noncompliance.
Unanticipated Problems/Serious or Continuing Noncompliance
The Committees on Human Research promptly report all unanticipated problems involving risks to subjects or others, serious or continuing noncompliance with applicable regulations or requirements of the IRB, and suspension or termination of IRB approval to appropriate institutional officials (the UVM Vice President for Research and the UVM Medical Center CMO when applicable) and federal agencies (i.e., OHRP, FDA and/or other agencies as appropriate). All such matters are appropriately reviewed, and any necessary actions are taken to ensure continued protection of human subjects.
Determination of Significant vs Non-significant Risk Devices
For research involving investigational devices, it is the responsibility of the convened Committee on Human Research in the Medical Sciences to determine which device studies pose significant or non-significant risk when the studies do not have an Investigational Device Exemption (IDE). Studies which are determined to be significant risk must obtain an IDE from the FDA. Studies which have already obtained an IDE are assumed to be significant risk studies and this determination is not necessarily discussed or documented.
IRB Record Requirements (Sec. __.115)
The IRB keeps all records in accordance with all pertinent regulations. This record keeping includes the following.
Membership rosters Sec. __.108(a)(2)The institution is required to maintain a current list of IRB members identified by name; earned degrees; representative capacity; indications of experience such as board certifications or licenses sufficient to describe each member's chief anticipated contributions to IRB deliberations; and any employment or other relationship between each member and the institution, for example, full-time employee, part-time employee, member of governing panel or board, stockholder, paid or unpaid consultant. UVM rosters indicate regular voting versus alternate members, specific OHRP designation, if applicable, as well as alternate replacement assignments. Rosters are updated each time there is a change in the Committee membership. Copies of curriculum vitae are obtained and kept on file for all primary and alternate members.
Written procedures and guidelines including, but not limited to, the IRB Policy and Procedure Research Manual, the FWA, and all website content.
IRB Minutes – see section 1.3.5
Convened CHRMS II and convened safety meeting minutes will be uploaded along with the agenda for the next full committee meeting for members to review online. Subsequent protocol reviews, just as amendments and continuing reviews will be reviewed through the CHRMS system of operations.
A report of business conducted by the expedited review process is available in the UVMClick-IRB system.
Protocol files as of 2017 are electronic. Any protocol materials received prior to that are in paper format. Both the paper and the electronic files include protocols, continuing reviews, modification, safety reports, adverse events, and consent documents. Once closed, any paper files are stored off-site and can be retrieved within 24-48 hours. Protocol files, whether paper or electronic will be destroyed after the protocol has been completed for at least six years.
Statements of significant new findings provided to subjects are kept in the protocol file.
Communications to and from the IRB are maintained in the protocol file.
Emergency use reports are kept in an electronic file.
Budget and accounting records when relevant.
Initial and Continuing Review (Sec. _115(a)(3) IRB will document decisions to require continuing review or full board review even in circumstances when such review is not required.
Single IRB (Sec._115(a)(9) IRB will maintain adequate documentation of the responsibilities that each entity will undertake to ensure compliance with the cooperative research policy.
Documentation for Full Committee Review For each protocol reviewed by the convened IRB, the following will be available in UVMClick:
o IRB Actions/determinations
o Review type
o Action
o The vote on IRB actions, including the number of members voting for, against, abstaining, and recusal
o A written summary of the discussion and resolution of controversial issues
o Minor or substantive revisions required to secure approval and the basis for the revisions
o The basis for disapproving research
o The effective date, the initial approval date and expiration date
Documentation of Actions Completed Outside of Convened Meetings For protocols approved with specific minor conditions, the minutes of the first IRB meeting that takes place after approval was finalized will provide the date the conditions were met.
o IRB minutes include sufficient information to notify IRB members of the following:
o Expedited reviews of new projects, including Expedited Review Category
o Expedited reviews of minor amendments to full board protocols, including a list of the amendments and confirmation the amendments met the criteria for Expedited Review (i.e., the amendments pose minimal risk)
o Expedited reviews of amendments to research initially approved by expedited procedures, including a list of the amendments and Expedited Review Category
o Expedited continuing reviews, including Expedited Review Category
o NOTE: For research reviewed under expedited procedures, regulatory requirements for discussions, decisions and findings (including protocol-specific justifications) will be documented UVMClick. This includes findings related to level of risk and waivers or alterations of informed consent.
Authority to Review/Sign IRB Documents
The IRB Chair and his/her designee(s) are authorized to sign any and all documents on behalf of the IRB in connection with the review and approval (or determination of exemption) of research project involving human subjects. Implementation shall be the responsibility of the RPO Director. All Member signatures are electronic and in compliance with FDA 21 CFR Part 11.
a. Results of Reviews, Actions and Decisions from a Full or Expedited Review
Depending upon the nature of the required conditions, the IRB designates any of the following individuals or groups of individuals to determine that the conditions of approval have been satisfied:
· The IRB chair or Associate Chair
· Another IRB member
· An IRB Regulatory Analyst or
Review of response materials from investigators requiring medical, scientific, or other technical expertise will be assigned to scientific members as applicable.
Response materials that do not require medical, scientific, or other technical expertise may be assigned to non-scientific members as applicable.
b. Specific authority granted to IRB Regulatory Analyst members using expedited review procedures
· Review and approval of exemption determinations
· Waiver and/or Alteration of HIPAA determinations
· Amendments that are administrative in nature such as
o New recruitment materials
o Change in sponsor
o Closure to accrual
o Change in collaborating sites
o Change in study title
o Continuing review of projects that are in data analysis, long term follow-up with no more than minimal risk procedures, protocols where work has not yet begun or grant proposals lacking definite plan for involvement of human subjects
o Change to previously approved recruitment material
o Correction of typographical and spelling errors in consent or protocol
o Correction of omission of sponsor
o IDB Updates, DSMB reports, Annual IDE Reports
o Updates to Data Management and Security Plans
o Changes to Key Personnel
o Retention materials (sponsor newsletters, calendars, water bottles etc.)
o Sponsors close out documents (thank you letters to patients and/or physicians, certificate of site completion.
c. Routine Internal Correspondence
Any action, letter, memo or e-mail between the Committee or IRB Regulatory analyst and the faculty or staff of the University that provides information concerning the review of research protocols by the Committee or IRB Regulatory analyst and which do not imply or appear to imply approval of this activity may be signed by an IRB Regulatory analyst.
Any letters, memos or email sent representing the decision or opinions of the Chairs of the IRBs or their respective designees, as long as such correspondence does not imply review and approval by the designee, may be signed by IRB Regulatory Analysts. (e.g., RNI, compliance memos)
Electronic Reviews
All reviews, initial, continuing reviews and modifications are completed electronically by the IRB members as assigned. Members receive an email notice that a review is pending. Members are required to authenticate into the electronic system using their UVM NetID and password prior to completing their review. The system validates the member’s authentication credentials based upon the member’s role in the system and determines available actions for each person. The electronic review is stamped within the system with the name of the individual carrying out the review activity (electronic signature), and the time and date that the electronic signature was applied to the review.
Members should only access records they have been assigned to review.
1.3.1 Medical Safety Subcommittee Review
Revised 6/5/23
Subcommittee Charge
The IRB is required to submit to the Office for Human Research Protections (OHRP) and the Food and Drug Administration (FDA) any unanticipated problems involving risk to subjects or others or any serious or continuing noncompliance [45 CFR 46.108(a)(4)(i) & 21 CFR 56.108 (b)(1)]. The Medical Safety Subcommittee (SSC) (and/or their delegate) is charged with reviewing Reportable New Information (RNI’s) submitted through the UVMClick system to identify those cases where additional reporting and protocol modifications may be required. The SSC is composed of a subset of both Medical & Behavioral and Social Science IRB Members, IRB Chairs, the RPO Director, the IRB Director and IRB regulatory analysts. RNI’s not of a medical nature are reviewed and discussed through the Behavioral and Social Sciences Full Committee (CHRBSS).
Initial Process for Review of Reportable New Information
RNI’s are submitted to the IRB electronically by the PI, the PI’s designee, or an independent reporter. The IRB Regulatory Analyst (as the Subcommittee delegate) makes an initial decision as to whether the RNI requires further review. The analyst may make an inquiry to the researcher for additional information, or they may consult with another Committee Member in the case of an adverse event for which medical expertise is necessary to make the determination. If the RNI is a minor deviation or other event that does not affect the safety and well-being of the participant or others, the report will be acknowledged via a memorandum back to the PI explaining the outcome of the review and that no further action is required.
Other RNI’s will be assigned to the next available SSC or CHRBSS agenda as New Business. If there is any doubt about the determination, the RNI will be placed on the agenda for discussion. The IRB Regulatory Analyst will electronically assign one SSC Member (primary) reviewer in UVMClick to present the RNI to the committee and make an initial recommendation. All Members will review the electronic agenda which will have embedded links to corresponding material such as the RNI submission, the last signed consent form, consent process documentation, physician notes or proposed corrective actions as presented by the PI.
Convened Safety Subcommittee or CHRBSS Process for Review of Reportable New Information
The primary reviewer and all committee members will have access to a review checklist partially completed by the IRB Regulatory Analyst and may be completed by the primary reviewer. The primary reviewer will summarize the issue, the proposed corrective action and will provide a preliminary recommendation.
The Committee will review and categorize the RNI as one of the following (may or may not be in UVMClick):
- Additional information required before deciding on a determination
- Unanticipated problem involving risks to subjects or others
- Non-compliance that does not impact rights or welfare (minor)
- Potential serious non-compliance (refer to Section 27 Noncompliance Policy)
- Potential continuing non-compliance (refer to Section 27 Noncompliance Policy)
- Other
The Committee will decide what if any further actions are required to protect research subjects. If further actions are required, a memorandum with the Safety subcommittee’s request is forwarded to the PI. This will be documented in the safety minutes. The safety minutes are available to the CHRMS Full Committee each month as part of the agenda for the next month’s convened meeting.
Actions when the RNI is an Unanticipated Problems Involving Risk to Subjects or Others
- The UAP will be placed on the next CHRMS or CHRBSS Full Committee agenda for review.
- The PI may be asked to voluntarily hold further accrual while the case is being reviewed.
- The Full Committee may require protocol/consent revisions and/or suspend or terminate the protocol.
- The Full Committee will report the UAP to the Institutional Official(s) and federal offices as applicable.
Actions when the RNI is Minor Noncompliance Not Impacting Rights or Welfare
Minor noncompliance that does not impact the rights or welfare of the participant will be informally resolved by the following steps in Section 27.1. Noncompliance Review Procedures
Actions when the RNI May Represent Serious or Continuing Noncompliance
Noncompliance that may be serious or continuing will be referred for review under 27.1 Noncompliance Review Procedures.
1.3.2 Guidelines for Continuing Review of Research
Revised 11/03/22
Federal regulations require an IRB to conduct substantive and meaningful continuing review of human subject’s research. Although an IRB may become familiar with various individual aspects of a research project’s conduct, the continuing review provides an opportunity to reassess the totality of the project and assure that, among other things, risks to subjects are being minimized and are still reasonable in relation to anticipated benefits, if any, to the subjects and the knowledge that is expected to result.
Both HHS and FDA regulations set forth the criteria for IRB approval of research. These criteria apply to both initial and continuing review of research and provide the framework for the IRB’s evaluation of research. The IRB must determine that all of the following requirements are satisfied:
- Risks to subjects are minimized (i) by using procedures which are consistent with sound research design, and which do not unnecessarily expose subjects to risk, and (ii) whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes (45 CFR 46.111(a)(1));
- Risks to subjects are reasonable in relation to anticipated benefits, if any, to the subjects and the importance of the knowledge that may reasonably be expected to result (45 CFR 46.111(a)(2));
- Selection of subjects is equitable (45 CFR 46.111(a)(3));
- Informed consent will be sought from each prospective subject or the subject’s legally authorized representative, and appropriately documented in accordance with, and to the extent required by, HHS regulations at 45 CFR 46.116 and 46.117, respectively (45 CFR 46.111(a)(4) and (5));
- When appropriate, the research plan makes adequate provision for monitoring the data collected to ensure the safety of subjects (45 CFR 46.111(a)(6));
- When appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data (45 CFR 46.111(a)(7));
- Appropriate safeguards are included to protect subjects likely to be vulnerable to coercion or undue influence (45 CFR 46.111(b)); and
- When the research involves pregnant women, fetuses, or neonates; prisoners; or children, the research satisfies the additional requirements for IRB approval under HHS regulations at subpart B, C, or D, respectively, of 45 CFR part 46.
Which Protocols Require Continuing Review
With OHRP’s 2018 Common Rule change, continuing review is no longer required for some minimal risk research, however, all FDA supported projects continue to require continuing review. See Section 16.0 for additional information.
Level of IRB Review
A protocol that requires continuing review may be reviewed at one of two levels:
Full Committee Review: Human research which does not meet the criteria for expedited review or exemption from IRB review must be reviewed by the Full Committee at a convened meeting. Continuing review of a protocol which initially required Full Committee review will continue to be reviewed by the convened Board unless:
- where (i) the research is permanently closed to the enrollment of newsubjects: (ii) all subjects have completed all research-related interventions; and (iii) the research remains active only for long-term follow-up of subjects; or,
- where no subjects have been enrolled and no additional risks have been identified; or
- where the remaining research activities are limited to data analysis; or
- the protocol is a Humanitarian Use Device protocol (see FDA Guidance document issued July 8, 2010)
Expedited Review: Research which meets the criteria for expedited review is reviewed by the Chair or his/her designee. A protocol which initially was reviewed using expedited review procedures may be reviewed for continuing review using expedited review procedures. However, research protocols that previously met the criteria for expedited review will require Full Committee review if changes to the protocol are proposed which: (1) present more than minimal risk to human subjects or (2) involve procedures which do not meet the criteria for expedited review.
Assigning and completing reviews
IRB staff will electronically assign the continuing review based on the level of review that is required. Expedited continuing reviews are assigned and completed by one member reviewer. Continuing reviews requiring Full review will be sent to one primary reviewer prior to the Full meeting where that protocol will be discussed. Continuing reviews will appear on the agenda, which is sent to the IRB Committee by IRB staff prior to the meeting so that they have time to view materials. The Committee members have available to them the same information as the Primary reviewer as well as to any comments made by the Primary reviewer once their review is complete. Ballots will be provided to Committee members present at the Full meeting and votes will be cast on the ballot which is then collected by the IRB staff.
As part of the electronic continuing review submission, the IRB member will review:
- Completed continuing review eform in UVMClick
- last signed consent form or addendum (if applicable) If more than one type of consent, all last signed consent should be uploaded for review. Participants names should be obscured allowing the date signed to be viewable.
- Any relevant monitoring visit reports from sponsors, auditors, or any regulatory body during the last year.
- Attach a list of non-risk deviations.
- a member reviewer checklist which has been partially completed by the IRB Regulatory Analyst to be verified by the member.
The member reviewer will have available to them the full protocol and all previous submissions and correspondences pertaining to the protocol electronically.
Note Regarding Non-Local Safety Reports: For research studies subject to oversight by a Data Safety and Monitoring Board (DSMB)/Data Monitoring Committee (DMC), the IRB will rely on current reports from the DSMB/DMC in lieu of reviewing non-local safety reports. The DSMB/DMC reports should include i) what information was reviewed by the DSMB/DMC, ii) the date of review, and iii) a summary of findings and/or recommendations. It is a requirement that DSMB/DMC reports be submitted to the Committee at least annually at time of continuing review.
Important issues for primary reviewers are:
- That the risks to subjects are still minimized.
- by using procedures which are consistent with sound research design, and which do not unnecessarily expose subjects to risk, and
- whenever appropriate by using procedures already being performed on the subjects for diagnostic or treatment purposes.
- That the risks to subjects are still reasonable in relation to anticipated benefits.
- That the number of subjects initially requested and approved has not been exceeded.
- Any protocol revisions that have been approved by the IRB since the last continuing review.
- Any request for protocol revision at the time of continuing review have been submitted.
- Determine if the study appears to be progressing as planned.
- If unexpected events, toxicity, or complications have occurred that may indicate a need for a change in the protocol or consent.
- If the subjects registered any complaints about the study.
- Whether the consent document that is currently in use contains all previous revisions.
- Review a current data safety and monitoring report to determine that the data and study events are being evaluated on a regular basis. It is appropriate for the IRB to require a protocol revision to improve the plans for data and safety monitoring if the IRB thinks this will improve the protection of research subjects.
Reviewer Responsibilities
- Review as above.
- Talk directly with PI to attempt to clarify/resolve major concerns prior to approval.
- Identify any remaining issues, which may need to be discussed further with the full Committee.
- Reviews are assigned electronically and should be completed electronically using the Electronic Continuing Review Instructions located on the Committee Login page.
1.3.3 Guidelines for Review of Modifications
The IRB is responsible for review of any changes to previously approved research prior to implementation. These changes are often referred to as “modifications” or “protocol revisions.”
Major modifications that potentially affect the risk/benefit ratio must be reviewed through the full committee review process, minor modifications not affecting the risk to subjects may be reviewed through the expedited review process.
When modifications impact the safety of subjects previously enrolled who continue to receive study interventions, it may be necessary to convey this information (e.g., to obtain the consent of the subjects) by means of an addendum to the existing consent form, use of a new form, or providing the subjects with an informational sheet regarding the update. The IRB may determine, for example, that such subjects must be notified of new findings or toxicities not noted at the time they were originally consented. Such notification is consistent with the view of informed consent as a continuous process, and affords subjects the opportunity to determine whether or not they wish to continue their participation in the research. The IRB shall determine on a case-by-case basis when such notification, and its documentation, is required.
The criteria for reviewing a modification are the same as those listed for initial review. However, in most cases the review of the modification can be more focused.
When a modification requires full review:
Member Reviewer should:
- Review the modification to determine;
- Does the revision affect the risk to subjects? If so, the following issues should be addressed: are the risks still reasonable in relation to the anticipated benefits and the importance of knowledge that may reasonably be expected to result? Does the protocol still meet the criteria that are used to evaluate new studies?
- Does the currently approved consent form require revisions to convey the potential risks? The modification should be accurately reflected in the consent form, if appropriate. Should currently enrolled subjects be informed or re-consented?
- Talk directly with PI to attempt to clarify/resolve major concerns prior to the meeting.
- Identify any remaining issues which need to be discussed.
- Ensure that the Lay Summary/Consent Form accurately and appropriately reflects the protocol and would be understandable to an average person.
- Outline any problems or issues that deal with the protocol or Consent Form. Editorial recommendations and indications of jargon can be corrected on your copy, submitted to staff and noted in the presentation that "editorial changes are suggested on my copy."
When modification requires expedited review:
Chair or designee should:
- Review as above.
1.3.4 Conducting IRB Business in the Event of a Pandemic or other Significant Emergency
Revised 05/20/20
Regardless of external events, the Office for Human Subjects Research Protections (OHRP) under DHHS expects each institution’s IRB to continue to conduct business according to the Common Rule, PHS Policy, and FDA requirements.
IRB Meetings
Regulations state that a convened meeting with a quorum present must conduct the following: 1) initial review of more than minimal risk protocols; 2) review of continuing studies where there is more than minimal risk; 3) any modifications that have the potential to increase risk; 4) determinations of level of seriousness for noncompliance cases. While optimal, there is no requirement to conduct the convened meeting in person. Use of teleconferencing or audio/video conferencing is permissible. If quorum cannot be achieved, convened meetings will be postponed until enough members can be present.
Security of IRB remote meetings will be assured by using only University-approved videoconferencing software logging in only with UVM credentials. Guest presence will be controlled by the meeting owner which, is typically an IRB staff person. IRB videoconference meetings will not be recorded. Quorum of members will assured by a count of those in attendance prior to opening the meeting and periodically throughout the meeting.
Minutes of meetings will be captured following current methods for in-person meetings. The manner of engagement of each member will be noted (e.g. in-person, telephone, video conference). Votes to go in or out of sessions, as well as to vote for specific protocols, will occur by the Chair asking for members who approve an order of business by asking “All approved say aye”, “All opposed say nay”, “All abstaining say aye”. Members participating through video conferencing can also use the chat feature to add comments to the discussion. Continuing review voting will be carried out by ballot and emailed to an IRB analyst . Members with conflicts will sign out of the meeting during the vote and IRB staff will invite them back into the meeting when the conflicting protocol discussion is complete. Meeting guests will be invited during discussion of their protocol and signed out once that discussion is completed.
Protocol Review
New, more than minimal risk, protocols, or amendments representing a potential for increase in risk, must be reviewed in a convened meeting. Convened meetings can proceed as described above. The CHRMS II Committee may be convened to review policies, protocols that have potential for benefit to individuals or to public health during a pandemic.
New, not more than minimal risk, protocols or amendments can continue to be reviewed through the current expedited process.
Protocol review documentation will be through the UVMClick-IRB electronic protocol submission software.
Quality Assurance Reviews
Scheduled quality assurance reviews will be placed on hold until normal working conditions are in place.
Pause on Human Subjects Research Activities
If human subject research activities are required to be placed on pause or altered to “avoid immediate hazard” secondary to institution-wide policy to address a public health situation, the IRB does not require notification in the pause of work. Alterations of protocol to avoid immediate hazard may be submitted at time of next annual progress report.
Education
Scheduled educational presentations and weekly in person drop in-hours will be placed on hold until normal working conditions are in place. Researchers are still encouraged to email RPO staff with protocol specific questions.
Training
If personnel refresher training expires during the emergency, personnel are allowed to continue their work as long as they have received previous training and have demonstrated proficiency. There will be no consequences; however, once working conditions have been restored, personnel must complete refresher training in accordance the IRB policy.
1.3.5 Institutional Review Board Minutes
Updated 11.02.22
Minutes of IRB meetings will be prepared in sufficient detail to demonstrate that IRB meetings were convened with a quorum of members and IRB deliberations reflect appropriate regulatory and local requirements. IRB Committee meeting minutes must accurately reflect the discussion and voting that took place at the meeting. IRB Regulatory analysts will take notes during attendance and subsequently prepare the minutes. The agendas and minutes of convened full-board IRB meetings will be maintained within the UVMClick system as well as stored on the UVM shared drive. The minutes will be reviewed and voted on at a future convened meeting then signed by the IRB Chair.
A separate report will include actions taken through expedited review procedures to ensure notification to all IRB members. This report is available monthly through UVMClick and upon request by IRB members.
Minutes include:
- Quorum: A record of quorum and/or loss of quorum at each IRB meeting, including presence of one member whose primary concern is in a non-scientific area.
- Minutes shall specifically note changes in the voting members present during voting on each item throughout the meeting, to document maintenance of quorum. This includes late arrivals and early departures.
- The minutes shall also note when departing members are replaced by other members during the meeting to maintain quorum.
- Attendance: A record of attendance of members, noting the key compositional requirements for quorum and noting which members are eligible to vote, and a record of attendance of guests at the IRB Committee meeting. Current member rosters with alternates will be appended to the minutes.
- Attendance by Alternate Means: A record of those members or alternate members who participated in the meeting in-person, through videoconference or teleconference (speakerphone),.
- Conflict of Interest: For any IRB Committee meeting in which an IRB Chair, Associate Chair or IRB member recuses himself/herself/themselves due to a conflicting interest with the research under review, the minutes reflect that a conflicting interest has been disclosed. The member with the conflicting interest may provide information on the item under review if requested by the IRB Chair or members. The minutes should reflect the member left the meeting and was not involved in the motion or the vote. The member is not counted towards quorum for that specific protocol discussion. Minutes must reflect the reason for the recusal as a conflict.
- Deliberations: Minutes will include an accounting for each item, including
- a summary of the discussion of controverted issues and their resolution,
- the basis for requiring changes in research, and
- the basis for disapproving research
- review and determinations of serious or continuing noncompliance – see section 27. Noncompliance Policy and Procedures
- Actions (Determinations): The minutes shall reflect actions and votes for each protocol undergoing initial review, continuing review, unanticipated problems, noncompliance, or review of modifications by the convened IRB. The IRB will document specific determinations and method of consent. A separate report will include actions taken through expedited review procedures to ensure notification to all IRB members. This report is available monthly through UVMClick and upon request by IRB members.
- Votes: A record of votes taken by the IRB Committee on all actions. This includes the total number of votes, the number of votes for, against, abstaining, and any recusals. If alternate members are voting that will be documented in the minutes as well. The vote on each action will reflect those members eligible to vote on that item.
- Review Period: For Initial and Continuing Review, a record of the duration of the approval granted to each protocol, as determined by the IRB.
- Vulnerable Populations: A record that reflects that the IRB reviewed additional safeguards to protect vulnerable populations if entered as study participants.
- Risk Level: The Committee will determine if a given protocol is high risk based on internal criteria and require additional oversight as felt necessary to protect human subjects.
- Security/Confidentiality/Protected Health Information (HIPAA): Security and storage of the data is reviewed by technical specialists and specific procedures and protections for protected health information (PHI) is recorded in the minutes.
- Consultants (and ad hoc Reviewers): If a consultant is present at the convened meeting, the name of the consultant, and a brief description of the consultant’s expertise will be documented. Additionally, it will be documented that the consultant was not allowed to vote.
- Required Justifications and Findings: There will be a description of any required findings the IRB must make along with the protocol-specific information justifying each IRB finding.
- Waiver or alteration of the consent process under criteria 45 CFR Section 116 or waiver of written documentation of consent under 45 CFR Section 117
- Research involving pregnant women, fetuses, and neonates under 45 CFR Subpart B
- Research involving prisoners, under 45 CFR C
- Research involving children under 45 CFR D
- FDA Regulations: The rationale for determining that risk associated with using a medical device in a study is significant or non-significant (referred to as significant risk/non-significant risk device determinations).
Review and Approval of Minutes
The draft minutes of each IRB meeting will be reviewed and voted on at the next available full Committee meeting. RPO staff distribute the full Committee meeting agenda, including the previous month’s minutes via email 5 days before the next meeting. This email is sent to all members, including those members unable to attend the upcoming meeting. All members review the minutes even if not in attendance to keep informed of IRB deliberations.
Regulations require we document those issues that are considered controverted, their resolution, basis for requiring changes and basis for disapproval. The minutes are not a verbatim transcript of everything said during a meeting. If a committee member does not feel the draft minutes represent what was discussed adequately, the original notes taken by the IRB Regulatory analyst will be re-reviewed by the Chair and IRB leadership and additional information included as appropriate. Information that was not discussed at the meeting cannot be included in the minutes. Discussions, emails, and phone calls that occur outside the convened meeting cannot be included as part of the minutes. Additional or new controverted issues will only be considered at a convened future meeting of all committee members.
Once approved by the members at a subsequent IRB meeting, minutes cannot be altered by anyone including a higher authority (except to correct any errors identified during quality review, in which case the minutes are subject to re-approval by the convened IRB). Minutes are available upon request to the Vice President for Research and federal agencies as part of auditing functions. Approved, signed minutes are retained in the UVMClick system and UVM shared drive for 6 years.
1.4 Public Records and Open Meetings (Vermont Law)
Revised 05/03/22
Federal Freedom of Information Act (FOIA)
The Freedom of Information Act (FOIA) is a federal law that generally provides that any person has a right, enforceable in court, to obtain access to federal agency records. All federal agencies, including EPA, are required to make requested records available unless the records are protected from disclosure by one of nine FOIA exemptions contained in the statute.
The FOIA applies only to federal agencies. It does not apply to records held by Congress, the courts, or by state or local government agencies. Each state has its own public access laws that should be consulted for access to state and local records. Vermont and in turn the University has its own laws and processes for records.
Vermont Public Records Act
The University of Vermont is a public body subject to the Vermont Public Records Act (1 V.S.A. §316) (a). Under this law, any person may inspect or copy any public record of a public agency. The definitions of public agency; public records and documents are included in 1 V.S.A. §317.
However, research data and protocols are exempt from disclosure under Vermont law §317(c)(23). Because these records are exempt from public disclosure, the FOIA cannot be used to inspect or copy records. Researchers should forward any request for inspection or copies of research records to the Vice President for Executive Operations according to the UVM Records and Document Request policy.
1.5 IRB Jurisdiction
New 05/23/21
All human subjects research conducted by UVM/UVMMC personnel and/or students fall under the jurisdiction of the UVM IRBs. For this guidance, these individuals are referred to collectively as a “UVM/UVMMC Work Force member.”
Categories that fall Under UVM IRB Jurisdiction
The following are the categories of human subjects research that must be reviewed by the UVM IRBs:
- The research is performed as an academic requirement for UVM/UVMMC employees or UVM students.
- The research is performed as part of an individual’s UVM scholarly activities.
- The research is conducted by a member during work/education/volunteer time or with UVM/UVMMC resources/money/space.
- The research is being performed as part of a UVM/UVMMC course or training program.
The following are some less obvious examples of when review by a UVM IRB is required:
- Member conducts research at a school, day care center, company, community center, or another healthcare facility.
- Member receives a grant/subcontract through UVM but the research is conducted by another institution.
- UVM data or samples are provided to external researchers for use at an offsite location.
- Member consults on the design of a research protocol to be conducted elsewhere and also participates in analysis of identifiable data.
Categories that do not fall Under UVM IRB Jurisdiction
Some human subjects research projects may not be eligible for review by UVM IRBs, although activities may be conducted by members of the UVM/UVMMC Work Force. The following are categories that do not require UVM IRB review:
- Members conducting research activities for an unaffiliated private practice/business/or other organization.
- Members conducting research to fulfill education requirements that are not associated with UVM academic requirements.
Note: If unaffiliated human subject research activities involve use of any resources from UVM/UVMMC, UVM IRB review is required.
When there is a question as to whether the UVM IRBs are the appropriate body to review the project, the researcher should contact the IRB administrative office. The IRB will consult with the appropriate Institutional Official to make this determination.
2.0 Institutional Ancillary Reviews
2. Institutional Ancillary Reviews
Revised 2.6.2023
Ancillary reviews provide the IRB with a method to allow protocol record access to authorized individuals, departments, offices, and other additional reviewers as needed, to provide feedback, approval, feasibility review etc., in parallel with the IRB review. Not all studies require ancillary review.
Applicability of an ancillary review is driven by federal, state and local regulatory requirements, whether UVMHN or UVM, while others are to ensure protocols meet local policy, are feasible or are simply for informational purposes only. Ancillary reviews can be assigned by the PI or their proxy or the IRB Analyst and are based upon the specifics of the protocol.
The ancillary reviewer will work directly with the PI regarding any clarifications or items that require resolution. These reviews can result in required changes to the protocol, consent or other materials as outlined by the ancillary reviewer. Once the submission is acceptable to them, you will be required to resubmit any revised documents back through the IRB system so the IRB reviewers have the most current version to conduct their review.
Each authorized entity has identified different stopping rules for their specific ancillary review process. Completion of an ancillary review may have the following impact on the review of your protocol through the IRB:
• no direct impact and is simply an FYI to the entity,
• it may be a condition of IRB approval release or
• IRB review may be held until the ancillary review is complete.
Below are examples of ancillary reviews that may be required during the protocol and/or modification review process.
Institutional Biosafety Committee (IBC)
Any IRB protocols that involve the use of recombinant DNA, gene therapy or biohazardous agents require review and approval, as mandated by the National Institutes of Health require review by the IBC. Approval to begin activities will not be released until IBC approval is obtained. Submissions may be made to both Committees simultaneously, but human subject activities must not begin until both Committees have approved the protocol.
Radiation Safety
The Radiation Safety Officer (RSO) at UVM oversees the use of ionizing radiation sources on UVM campus and ensures compliance with state and federal regulations, to protect UVM employees, students, the public, and the environment. Any protocols involving the application of radioactive materials, radioisotopes, and/or radiation treatment to humans for nonclinical purposes will undergo review by the UVM Radiation Safety Office when the research procedures are taking place at UVM.
If the research procedures are taking place in UVM Medical Center, the UVM Medical Center Radiation Safety Committee will review.
UVMHN Data Management Office (DMO)
Any medical record review protocols whether at UVMMC or any UVMHN affiliate requesting data from the DMO, must obtain approval from the DMO first. Once approved by the DMO, the IRB will review to ensure HIPAA regulations are met prior to release of Protected Health Information (PHI).
UVM Medical Center Billing Compliance Office
The IRB assists UVM Medical Center Compliance in initially identifying protocols that require an analysis and billing plan under Medicare requirements. IRB approval will not be released until we have been notified there is a final billing plan in place.
The University of Vermont Cancer Center Protocol Review and Management Committee (PRMC)
The PRMC reviews cancer-related protocols for scientific merit and progress, including participant accrual. It also prioritizes cancer protocols that may compete for the same patient population. Its function is complementary to that of the Institutional Review Board, which focuses on the protection of human subjects in research. Any initial review or modifications to protocols regarding the study of cancer require prior review by the PRMC. Review by the PRMC is independent of the review by the Institutional Review Board (IRB). PRMC and IRB, however, do share their committee review correspondence and outcomes with each other. While submissions may be made to both Committees at the same time, the PRMC must approve the protocol or approve with clarifications (that do not require changes to the protocol or consent or review at a subsequent full committee) prior to the IRB review. This is to ensure the PRMC clarifications and responses can be taken into consideration during the IRB review. This is required for all initial submissions and modifications.
Sponsored Project Administration (SPA)
Federal sponsors require documentation of IRB review prior to awarding funds to researchers. To ensure this requirement is met, IRB Analysts link protocols to grants as part of initial review and at time of funding source changes. SPA then checks to make sure there is an applicable protocol linked to their funding proposal prior to release of funds for sponsored projects.
Clinical Research Center Scientific Advisory Committee (SAC)
Any protocols wishing to utilize resources of the Clinical Research Center require review by the SAC. Review by the SAC is independent of the review by the Institutional Review Board (IRB). However, the findings from the IRB review are shared through the electronic system. Submissions may be made to both Committees at the same time; however, SAC will not approve until the IRB has approved the project.
UVMMC Investigational Drug Services (IDS)
Any protocol that includes administration of drugs not yet approved by the FDA for use or drugs being tested for an unapproved use, regardless of how the medications are dispensed, will be assigned an IDS ancillary review. This review will be to assess feasibility and plans for adherence to VT Board of Pharmacy rules as applicable. See UVMMC Pharm3 policy. IDS will need to review and approve a research study prior to IRB review.
UVMMC Infectious Disease Practice Committee (IDPC)
The IDPC is a subcommittee of the Pharmacy and Therapeutics Committee focused on antimicrobial stewardship within the hospital. IDPC review is required for any protocol that involves antimicrobials prior to IRB submission in UVMClick-IRB. Researchers must submit the protocol to the IDPC co-Chairs who will review and provide either an approval or denial of approval to conduct this work in the hospital. See Pharm3 hospital policy.
Research Integrity – Financial Conflicts of Interest
The Office of Research Integrity provides support to investigators, faculty, and staff with financial conflicts of interest in sponsored projects. The IRB has access to the conflict of interest disclosure database to establish whether there are significant financial conflicts of interest or a management plan that requires disclosure to potential research subjects. Ancillary reviews may be assigned as needed.
Office for Clinical Trials Research (OCTR) - Contracts and Invoices
OCTR supports researchers conducting industry- funded protocols by developing contracts, data use agreements on behalf of the hospital, invoicing and collecting IRB fees. OCTR also manages the credentialing process for University of Vermont clinical research personnel wishing to access hospital records for research purposes. Ancillary reviews are assigned for invoices, contracts and DUA’s.
Office of General Counsel
Rarely, the IRB will seek guidance for specific situations from either of the two institutional general counsel offices. Information about specific protocols may be shared. FOIA requests are always deferred to Risk Management and general counsel. Ancillary reviews may be assigned as needed.
Faculty Sponsor
Students meeting the definition as outlined in the IRB Policy and Procedures Manual must obtain their faculty sponsor review and approval through the UVMClick system prior to IRB review. An ancillary review will be assigned to the role of faculty sponsor in the UVMClick system.
Information Systems
The IRB has an IT professional on the IRB Committee to specifically assist us in determining appropriate measures for the protection of human subject data on the protocol level and an institutional policy level. Ancillary reviews may be assigned as necessary.
3. IRB Review Categories (Sec._46.109(a))
3.1 Full Committee Review
Revised 6.5.2023
The IRBs employ the convened meeting review process for review and approval of studies that are more than minimal risk.
Notification to Research Community
Committee meetings are noticed on the Committee website. Deadline for submission of new protocols is 4 weeks in advance. This time allows for the appropriate pre-review procedures as described below.
Notification to the Committee
Distribution of the Agenda to all members occurs approximately 2 weeks in advance of the convened meeting. Approximately 5 days prior a convened meeting a second distribution of any additional continuing full protocols received after the deadline is sent to all members. All new protocols, modifications, continuing reviews, and other business requiring full committee action are placed on the Agenda for discussion.
Access to the Protocol Materials
Committee members have access to all of the protocol materials through the electronic submission and review system.
Pre-review Procedures
Similar to granting agency review, IRBs must receive sufficient information from investigators to provide adequate review of proposed research and to make the determinations required by regulations for IRB approval.
Inclusion on a Committee agenda is not guaranteed, until your department-assigned IRB analyst completes a pre-review of the submission. A specific checklist is followed to determine if the submission is ready for full committee review. If the submission is incomplete or lacks information necessary to conduct a full committee review, it will be withdrawn from the current agenda. The PI will be notified along with a list of requirements for resubmission. It will be placed on an agenda when the submission is found to be satisfactory.
Reviewer Assignments
A Primary and Secondary Reviewer is assigned to review the complete protocol, consent form, Investigational Drug/Device Brochure and any other protocol materials. This includes any pre-review checklists completed by the IRB analyst. Efforts are made to match the primary reviewer’s expertise to the protocol subject matter. Reviewers are encouraged to contact the PI to resolve/clarify major concerns prior to the meeting. The reviewers summarize the protocol or amendment for the full Committee at a convened meeting and answer questions during the discussion. The member reviewer(s) will determine that the following requirements are satisfied prior to approval:
Requirements for Approval
1. Risks to subjects are minimized:
a. by using procedures which are consistent with sound research design;
b. do not unnecessarily expose subjects to risk, and
c. whenever appropriate by using procedures already being performed on the subjects for diagnostic or treatment purposes.
2. Risks to subjects are reasonable in relation to anticipated benefits to subjects, and the importance of knowledge that may reasonably be expected to result.
In evaluating risks and benefits only those risks and benefits that may result from the research should be considered (as distinguished from risks and benefits of therapies subjects would receive even if not participating in the research).
3. Selection of subjects is equitable. The purposes of the research, the setting in which the research will be conducted, and the population from which subjects will be recruited should be taken into account.
4. Absent the IRB granting a waiver of consent or waiver of documentation of consent, informed consent will be sought from each prospective subject or the subject’s legally authorized representative and will be documented in accordance with applicable regulations. (45 CFR 46.116&46.117) Consent document will be reviewed to ensure it accurately reflects the protocol and would be understandable to a reasonable person.
5. Where appropriate, the research plan makes adequate provisions for monitoring the data collected to ensure the safety of subjects.
6. Where appropriate, the research plan makes adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data.
7. Where some or all of the subjects are likely to be vulnerable to coercion or undue influence, such as children, prisoners, persons who lack capacity to consent, or economically or educationally disadvantaged persons, appropriate safeguards are included in the study to protect the rights and welfare of these subjects. (Subparts B, C, D)
Documentation
The IRB minutes will include documentation of the discussion of the studies that were reviewed at the convened meeting as well as the votes and any abstentions, recusals, and determinations of applicability of any subparts. The continuing review votes are accomplished by paper ballot in the meeting and entered into the electronic system post meeting.
Other Full Committee Actions
The full committee may also discuss and vote on new policies as needed, noncompliance investigations, and Unanticipated Problems Involving Risk to Subjects or Others.
3.2 Expedited Review (Sec._46.110)
Revised 5.3.2023
The IRBs employ the expedited review process for approval of studies that are determined to be minimal risk and only involves activities on the Secretary’s list (OHRP, FDA or other Federal Agency, as indicated in Code of Federal Regulations, ( 45 CFR 46.110 and 21 CFR 56.110).
To see a list of expedited categories, click here.
UVM’s IRB expedited review process is also currently employed for approval of
- study modifications involving no more than minimal risk,
- when conducting limited IRB review as required by the exemptions at Sec. _.104(d)(2)(iii), Sec. _.104(d)(3)(i)(C)
- Secondary research under 45 CFR 46.104(d)(4)(iii)
- when the IRB reviews and approves research with conditions at a convened meeting without requiring further review at a subsequent convened meeting,
- Some continuing reviews
Note: The IRB is required to document rationale when they override the presumption that studies on the Secretary’s expedited review list involve greater than minimal risk (Sec. _.115(a)(8)).
Pre-review Procedures
IRBs must receive sufficient information from investigators to provide adequate review of proposed research and to make the determinations required by regulations for IRB approval. Submission requirements can be found on our website and the IRB analyst can be contacted with any questions.
Review by a member is not guaranteed, until your department-assigned IRB regulatory analyst completes a pre-review of the submission. A specific checklist is followed to determine if the submission is ready for member review. If the submission is incomplete or lacks information necessary to conduct a member review, the PI will be notified along with a list of requirements for resubmission. The protocol will be sent for review when the submission is found to be satisfactory.
Reviewer Assignment
One Committee member is assigned to review the complete protocol or amendment, consent form, recruitment material and any other applicable protocol materials. This includes any pre-review checklists completed by the IRB regulatory analyst.
The designated IRB member reviewer will conduct the review and document his/her determination of the applicable limited or expedited review category. Consistent with review by a convened IRB, expedited member reviewer will consider the Requirements for Approval as listed above.
Expedited member reviewers, as designated by the IRB, may exercise all of the authorities of the IRB, except that he/she may not disapprove the research. A research proposal may be disapproved only after review by the convened meeting.
Notification to the Board
When the expedited review procedure is used, all regular members will have access to these actions via the Expedited Submissions Approved in the Last 45 Days report that is available in the meeting workspace in UVMClick-IRB.
3.3 Limited Review (Sec._46.104)
Revised 10/02/10
The limited IRB review assures adequate protections for the privacy of subjects and adequate plans to maintain the confidentiality of the data required by 46.111(a)(7).
This new provision for limited IRB review allows certain research to be categorized as exempt, even when the identifiable information might be sensitive or potentially harmful if disclosed.
When is Limited IRB Review Used
Limited review is triggered for exempt categories 2, 3, 7, and 8 when:
1. The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, AND
2. Any disclosure of the human subjects’ responses outside the research would reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, educational advancement, or reputation.
Exempt category 2(iii) Research that only includes interactions involving educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observations of public behavior (including visual or auditory recording.) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can be readily ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to make the determination required by 46.111(a)(7).
Exempt category 3(i)(c) Research involving benign behavioral interventions in conjunction with the collection of information from an adult subject through verbal or written responses (including data entry) or audiovisual recording if the subject prospectively agrees to the intervention and information collection and the information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to make the determination required by 46.117(a)(7).
Limited review does not need to be conducted if the identifiable data would not reasonably place the subjects at risk. UVM is currently not implementing the new Exemption categories 7 and 8 at this time. Continuing review is not required for research approved under limited IRB review.
Assuring Appropriate Protections
A completed Data Management and Security form is required. The information provided on this form will assist in the review of the following items:
• The nature of the identifiers associated with the data
• The justification for needing identifiers in order to conduct the research • Characteristics of the study population
• The proposed use of the information
• The overall sensitivity of the data being collected
• Persons or groups who will have access to study data
• The process used to share the data
• The likely retention period for identifiable data
• The security controls in place
o Physical safeguards for paper records
o Technical safeguards for electronic records
o Secure sharing or transfer of data outside the institution, if applicable
• The potential risk for harm that would occur if the security of the data was compromised.
Individuals Performing the Limited IRB Review
Limited IRB review must be performed by the IRB Chair or by an experienced IRB member. IRB analysts are IRB members and have the authority delegated to conduct these determinations. The review can occur on an expedited basis and does not require consideration by a convened board. The reviewer may require modifications to the proposal prior to approval. Disapprovals must be made by the convened board. If the limited IRB review does not result in approval under the exempt categories, then the IRB can evaluate whether or not approval is appropriate under the expedited categories. Expedited research must meet all the approval criteria under 45 CFR 46.111, including either informed consent or waiver of consent.
3.4 Exemption Determinations (Sec.__.104)
Revised 5/9/2023
Convened Committee review is not required for certain categories of research activities that involve little or no risk to human subjects. However, the University and UVM Medical Center have an obligation to be apprised of all human subjects’ research being conducted under their auspices in the event any questions or problems arise and to assure that, regardless of risk, all research subjects are afforded the same protection. Therefore, the UVM IRB will make all exempt determinations for UVM/UVMMC research projects.
Research which is determined to be exempt from Convened Committee review, must comply with all University of Vermont policies and procedures, as well as with applicable federal, state and local laws regarding the protection of human subjects in research. Please note, however, that although the research is exempt from formal review, it is not necessarily exempt from informed consent requirements.
Authority to Grant Exempt Status
The rule has modified some of the categories to now allow recording of identifiable sensitive information. Determinations in these modified categories 2(iii) & 3(i)(c) will require an IRB member to conduct a limited review. Protocols where protected health information is used or disclosed fall into exempt 4(iii) Secondary Research which requires an IRB member to conduct an expedited HIPAA review. IRB regulatory analysts are designated IRB members and will review these studies and make determinations.
Length of Determinations
Once a project has been determined to meet the criteria for exemption, there is no expiration date for the project and ongoing IRB oversight is not required.
Changes to the Project/Amendments
Any proposed changes to the project that may affect the original determination of exemption must be prospectively submitted for review and subsequent determination of exemption. Changes to a project initially determined exempt under Category 4(iii) Secondary Research always requires submission for a subsequent determination under the HIPAA Privacy Rule.
Exempt Research and Vulnerable Populations
§46 Subpart B —Pregnant Women and Human Fetuses Involved in Research
- The DHHS categories of exempt research §46.101(b)(1) through (6) may apply to pregnant women and fetuses or their records.
§46 Subpart C - Biomedical and Behavioral Research Involving Prisoners as Subjects
- The DHHS exempt categories do not apply to research involving prisoners.
§46 Subpart D and §46.104 - Exempt Research Involving Children
- With adequate protections for ensuring individual privacy and data confidentiality, research involving children, or their records may be exempt under DHHS categories 1, 2, 4, 5, and 6.
- Exemptions 2(i) and 2(ii) may apply to research with children involving educational tests or the observation of public behavior only if the investigators do not participate in the activities being observed.
- Exemptions 2(iii) and 3 (benign behavioral interventions) do not apply to research involving children.
Exempt Research Involving Adults with Impaired Decision-making Capacity
- With adequate protections for ensuring individual privacy and data confidentiality, research involving adults with impaired decision-making capacity may be exempt under DHHS categories 4 or 5.
- Research involving interactions or interventions with adults with impaired decision-making capacity cannot be reviewed as exempt.
Exempt Research Involving Non-English Speakers
- Projects involving interactions with non-English speakers may be eligible for exempt review under DHHS categories 1-3, and 5-6.
- For projects involving with non-English speakers, researchers must include mechanisms to ensure participants comprehend the study purpose, what participants will experience, participant risks (if any), and research benefits.
Exemption Categories
The exempt categories have been revised and expanded under the 2018 Common Rule change.
#1. EDUCATIONAL STRATEGIES, CURRICULA OR CLASSROOM MANAGEMENT METHODS 45 CFR 46.104(d)(1)
(1) Research, conducted in established or commonly accepted educational settings, that specifically involves normal educational practices that are not likely to adversely impact students' opportunity to learn required educational content or the assessment of educators who provide instruction. This includes most research on regular and special education instructional strategies, and research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods.
#2 SURVEYS, INTERVIEWS, EDUCATIONAL TESTS, AND OBSERVATION OF PUBLIC BEHAVIOR 45 CFR 46.104(d)(2)
(2) Research that only includes interactions involving educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior (including visual or auditory recording) if at least one of the following criteria is met:
(i) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained, directly or through identifiers linked to the subjects;
(ii) Any disclosure of the human subjects' responses outside the research would not reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects' financial standing, employability, educational advancement, or reputation; or
(iii) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to make the determination required by § __.111(a)(7).
#3 BENIGN BEHAVIORAL INTERVENTION 45 CFR 46.104(d)(3)
The 2018 Common Rule added this new exemption category because respect for persons is accomplished through the prospective subject’s forthcoming agreement to participate, the research activities pose little risk, and the use of this exemption for many social and behavioral studies will enable IRBs to devote more time and attention to studies involving greater risk or ethical challenges.
(3)(i) Research involving benign behavioral interventions in conjunction with the collection of information from an adult subject through verbal or written responses (including data entry) or audiovisual recording if the subject prospectively agrees to the intervention and information collection and at least one of the following criteria is met:
(A) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained, directly or through identifiers linked to the subjects;
(B) Any disclosure of the human subjects' responses outside the research would not reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects' financial standing, employability, educational advancement, or reputation; or
(C) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to make the determination required by § __.111(a)(7).
(ii) For the purpose of this provision, benign behavioral interventions are brief in duration, harmless, painless, not physically invasive, not likely to have a significant adverse lasting impact on the subjects, and the investigator has no reason to think the subjects will find the interventions offensive or embarrassing. Provided all such criteria are met, examples of such benign behavioral interventions would include having the subjects play an online game, having them solve puzzles under various noise conditions, or having them decide how to allocate a nominal amount of received cash between themselves and someone else.
(iii) If the research involves deceiving the subjects regarding the nature or purposes of the research, this exemption is not applicable unless the subject authorizes the deception through a prospective agreement to participate in research in circumstances in which the subject is informed that he or she will be unaware of or misled regarding the nature or purposes of the research.
#4 SECONDARY RESEARCH (IDENTIFIABLE PRIVATE INFORMATION/BIOSPECIMENS) 45 CFR 46.104(d)(4)
(4) Secondary research for which consent is not required: Secondary research uses of identifiable private information or identifiable biospecimens, if at least one of the following criteria is met:
(i) The identifiable private information or identifiable biospecimens are publicly available;
(ii) Information, which may include information about biospecimens, is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained directly or through identifiers linked to the subjects, the investigator does not contact the subjects, and the investigator will not re-identify subjects;
(iii) The research involves only information collection and analysis involving the investigator's use of identifiable health information when that use is regulated under 45 CFR parts 160 and 164, subparts A and E (HIPAA), for the purposes of “health care operations” or “research” as those terms are defined at 45 CFR 164.501 or for “public health activities and purposes” as described under 45 CFR 164.512(b); an IRB conducts a expedited HIPAA review to make the determination;
(iv) The research is conducted by, or on behalf of, a Federal department or agency using government-generated or government-collected information obtained for nonresearch activities, if the research generates identifiable private information that is or will be maintained on information technology that is subject to and in compliance with section 208(b) of the E-Government Act of 2002, 44 U.S.C. 3501 note, if all of the identifiable private information collected, used, or generated as part of the activity will be maintained in systems of records subject to the Privacy Act of 1974, 5 U.S.C. 552a, and, if applicable, the information used in the research was collected subject to the Paperwork Reduction Act of 1995, 44 U.S.C. 3501 et seq.
#5 PUBLIC BENEFIT/SERVICE PROGRAM RESEARCH (FEDERAL DEMONSTRATION PROJECTS)
45 CFR 46.104(d)(5)
(5) Research and demonstration projects that are conducted or supported by a Federal department or agency, or otherwise subject to the approval of department or agency heads (or the approval of the heads of bureaus or other subordinate agencies that have been delegated authority to conduct the research and demonstration projects), and that are designed to study, evaluate, improve, or otherwise examine public benefit or service programs, including procedures for obtaining benefits or services under those programs, possible changes in or alternatives to those programs or procedures, or possible changes in methods or levels of payment for benefits or services under those programs. Such projects include, but are not limited to, internal studies by Federal employees, and studies under contracts or consulting arrangements, cooperative agreements, or grants. Exempt projects also include waivers of otherwise mandatory requirements using authorities such as sections 1115 and 1115A of the Social Security Act, as amended.
(i) Each Federal department or agency conducting or supporting the research and demonstration projects must establish, on a publicly accessible Federal Web site or in such other manner as the department or agency head may determine, a list of the research and demonstration projects that the Federal department or agency conducts or supports under this provision. The research or demonstration project must be published on this list prior to commencing the research involving human subjects.
#6 TASTE/FOOD QUALITY EVALUATION & CONSUMER ACCEPTANCE
(6) Taste and food quality evaluation and consumer acceptance studies:
(i) If wholesome foods without additives are consumed, or
(ii) If a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture.
#7 STORAGE / MAINTENANCE OF IDENTIFIABLE DATA/BIOSPECIMENS OBTAINED WITH "BROAD CONSENT" (NEW) 45 CFR 46.104(d)(7)
UVM will not implement Exemption #7 at this time.
#8 USE OF IDENTIFIABLE DATA/BIOSPECIMENS OBTAINED WITH "BROAD CONSENT" (NEW)
45 CFR 46.104(d)(8)
UVM will not implement Exemption #8 at this time.
Notification
Exemption determination documentation is forwarded to the principal investigator indicating the exemption category.
3.5 Research Not Involving Human Subjects
Revised 6.5.2023
Projects that are not considered research and do not involve human subjects as defined by the regulations under 45 CFR 46.102(e)(1) do not require submission to the IRB for review.
Definitions
OHRP Definition of Research: a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.
A systematic investigation is an activity that involves a prospective plan to obtain data and conduct an analysis to answer a question. Typically includes:
- interventional medical procedures,
- surveys and questionnaires, interviews, and focus groups
- observational studies
- analysis of existing data or biological specimens
- evaluations of social or educational programs
- medical chart review studies
Generalizable knowledge is information that expands the knowledge base of a scientific discipline or other scholarly field of study. If the results of the systematic investigation are expected to be generalized to a larger population beyond the site of the data collection and replicated in other settings, then the knowledge is generalizable.
OHRP Definition of Human Subject: a living individual about whom an investigator (whether professional or student) conducting research
- Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or
- Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
FDA Definition of Human Subject: individual who is or becomes a participant in research, either as a recipient of a test article or as a control. A subject may be either a healthy human or a patient.
Intervention: includes both physical procedures by which data are gathered (for example, venipuncture) and manipulations of the subject or the subject's environment that are performed for research purposes.
Interaction: includes communication or interpersonal contact between investigator and subject.
Private Information includes information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public (for example, a medical record).
Identifiable private information is private information this is individually identifiable for which the identity of the subject is or may readily be ascertained by the investigator or associated with the information.
Identifiable biospecimen is a biospecimen for which the identity of the subject is or may readily be ascertained by the investigator or associated with the biospecimen.
De-Identified (HIPAA): Information that does not identify an individual and with respect to which there is no reasonable basis to believe that the information can be used to identify an individual. This includes that there is no means to re-identify individuals after the data have been de-identified (e.g. using a code or other means of record identification).
Readily identifiable does not mean potentially identifiable or identifiable with substantial effort.
Who is a Human Subject
Under regulation 45 CFR 46.102 (e), the definition of a human subject is
(1) Human subject means a living individual about whom an investigator (whether professional or student) conducting research
(i) Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or
(ii) Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
Who is Not a Human Subject
The regulations do not provide a definition, but these are common scenarios where individuals are involved in research, but their involvement does not fit the definition of a human research subject.
Decedents: Definition of human subject includes the requirement to be “living individuals”.
Individuals Not Readily Identifiable: De-identified data and individuals who are not readily identifiable are not human subjects. A dataset may contain HIPAA identifiers but might still not be readily identifiable. For example, data obtained from the PHIS dataset or Medicaid is not considered readily identifiable even though there are birth dates because the data comes from the entire nation.
Inanimate Objects: The subject of the research could be about institutions, programs, or hospitals and not about the individuals who are in those programs. For example, a survey could inquire about a training program, hospital or other institution and requiring information that doesn't contain information about individuals. In this case, even though a person fills out the questionnaire, the research is not about them as individuals - it’s about the program in which they work.
Deceased Individuals
Deceased individuals do not meet the definition of “human subjects” under the regulations. No IRB review is necessary, however, there are regulations regarding use of their protected health information. Investigators need to work with the hospital privacy officer to allow for use of the decedent’s protected health information. The required hospital form, “Attestation Form for Decedent Research” is on our forms webpage.
Self-Determination of Projects not Requiring IRB Review
It can be difficult to determine whether and which identifiers could readily identify an individual, but it is important to determine whether you are conducting research with human subjects so that you meet your human subject regulatory requirements. To assist researchers with this determination, the UVM IRB has developed a Qualtrics self-determination survey tool. The on-line tool will provide researchers with self-guided questions to determine if IRB review is required or the project is not considered research and does not involve human subjects as defined by the regulations under 45 CFR 46.102(e)(1) The tool can be found here.
3.6 Projects Not Requiring IRB Review
Revised 3.22.2023
IRB review is required when an activity constitutes human subjects research. Quality Improvement (QI) and Quality Assurance (QA) projects always involve human subjects, but only have to be submitted to the IRB for review if they meet the definition of research. Determining whether a project constitutes human subjects research rather than quality assurance, quality improvement, program evaluation or a public health practice involves multiple factors. The federal definition of research is “a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.” In contrast, QA/QI projects are typically designed, or intended, to:
- improve patient care;
- compare a program/process/system to an established set of standards such as standard of care, recommended practice guidelines, or other benchmarks;
- improve the performance of institutional practice or local systems;
- bring about improvements in health care delivery.
The projects are not designed to contribute to, or to advance generalizable knowledge. Instead, they are designed to develop or contribute to knowledge relevant to the organization. See the table below for a summary of the differences.
If any QA/QI project meets the criteria for research and involves human subjects, prior IRB review is needed. Regardless of whether IRB review is needed, all Protected Health Information must be transmitted, stored, analyzed, or otherwise exist only on HIPAA-compliant UVM/UVMH controlled electronic systems that meet security standards for protection of PHI.
Determinations of Projects Not Requiring IRB Review
The IRB Office is often requested to provide a determination on whether a project is research under the Federal regulations. Formal IRB determinations are requested in anticipation of such documentation being required for journals, conferences, funding sources and others. The IRB has developed a self-determination tool which guides researchers through a series of questions designed to determine if their project should be submitted to the IRB for review and approval.
Documentation of Projects Not Requiring IRB Review
Documentation can be printed at the completion of the self-determination tool to keep for your records and for any journal requests. The IRB will maintain a database of these submissions for quality checks
Proceed to the Self-Determination Tool
Overview of Differences between Research Under Regulations and Quality Improvement or Program Evaluation
| Research | Quality Improvement | Program Evaluation |
|---|---|---|---|
Intent/Purpose | Intent of project is to develop or contribute to generalizable knowledge (e.g., testing hypotheses) | Intent of project is to improve a practice or process within a particular institution or ensure it confirms with expected norms | Intent of project is to improve or assess a specific program |
Deviation from Standard Practice | May involve significant deviation from standard practice | Unlikely to involve significant deviation from standard practice | |
Design | may involve randomization of individuals to different treatments, regimens or educational practices | Generally does not involve randomization to different treatments, or practices. | Does not involve randomization of individuals, but may involve comparison of variations in programs |
Effect on Program or Practice Evaluated | Findings of the study are not expected to directly affect institutional or programmatic practice | Findings of the study are expected to directly affect institutional practice and identify corrective action(s) needed | Findings of the evaluation are expected to directly affect the conduct of the program and identify improvements |
Population | Usually involves a sample of the target population- universal participation of an entire clinic, program, or department is not expected; statistical justification for sample size is used to ensure endpoints can be met | Information is collected on all or most of the target population. | Information can be obtained from just a sample to all of the participants in the program |
Risks/Burdens | May put participants at risk | Does not increase risk to participants, with exception of possible privacy or confidentiality concerns | No risks to participants expected |
Dissemination of Results | Intent to publish or present generally presumed.
| If the results are publicized, they are described as “quality improvement” in public presentations, academic curriculum vitae, publications, etc. | Intent to disseminate the information to program stakeholders and participants is assumed This may be publicly posted (e.g., website or journal publications) to ensure transparency of results. |
According to DHHS guidance, “the intent to publish is an insufficient criterion for determining whether a QI activity involves research.”
The 2018 Common Rule change, effective 1/21/19, has specifically deemed a few activities as not research. See below:
(1) Scholarly And Journalistic Activities (E.G., Oral History, Journalism, Biography, Literary Criticism, Legal Research, And Historical Scholarship) (§ __.102(L)(1))
Scholarly and journalistic activities (e.g., oral history, journalism, biography, literary criticism, legal research, and historical scholarship), including the collection and use of information, that focus directly on the specific individuals about whom the information is collected.
(2) Public Health Surveillance (§ __.102(L)(2))
Public health surveillance activities, including the collection and testing of information or biospecimens, conducted, supported, requested, ordered, required, or authorized by a public health authority. Such activities are limited to those necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance (including trends, signals, risk factors, patterns in diseases, or increases in injuries from using consumer products). Such activities include those associated with providing timely situational awareness and priority setting during the course of an event or crisis that threatens public health (including natural or man-made disasters).
(3) Criminal Justice Activities (§ __.102(L)(3)) And Authorized Operational Activities In Support Of National Security Missions (§ __.102(L)(4))
Collection and analysis of information, biospecimens, or records by or for a criminal justice agency for activities authorized by law or court order solely for criminal justice or criminal investigative purposes.
(4) Authorized Operational Activities In Support Of National Security Missions
Authorized operational activities (as determined by each agency) in support of intelligence, homeland security, defense, or other national security missions.
Examples of implementing a practice and collecting patient or provider data for non-research clinical or administrative purposes include:
A radiology clinic uses a database to help monitor and forecast radiation dosimetry. This practice has been demonstrated to reduce over-exposure incidents in patients having multiple procedures. Patient data are collected from medical records and entered into the database. The database is later analyzed to determine if over-exposures have decreased as expected.
A group of affiliated hospitals implements a procedure known to reduce pharmacy prescription error rates and collects prescription information from medical charts to assess adherence to the procedure and determine whether medication error rates have decreased as expected.
A clinic increasingly utilized by geriatric patients implements a widely accepted capacity assessment as part of routine standard of care in order to identify patients requiring special services and staff expertise. The clinic expects to audit patient charts in order to see if the assessments are performed with appropriate patients and will implement additional in-service training of clinic staff regarding the use of the capacity assessment in geriatric patients if it finds that the assessments are not being administered routinely.
3.6.1 Case Studies
Case studies typically involve the collection of existing data and presentation of detailed information about a particular patient/person or small group to highlight an interesting condition, treatment, presentation or outcome.
A case report is a medical/educational activity that does not meet the DHHS definition of “research”, which is: "a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge." Therefore, the activity does not have to be reviewed by the IRB.
Under HIPAA, a case report is an activity to develop information that is shared for medical/educational purposes. Although the use of protected health information to prepare the paper does not require IRB review, the author of a case report must comply with HIPAA. Questions about HIPAA should be directed to the UVMMC HIPAA Privacy Specialist.
Investigators must also keep in mind professional requirements to obtain permission from the individual prior to collection of their data for this purpose.
3.7 Determination of Institutional Engagement in Research
Researchers are not always clear as to whether they or their colleagues are engaged in the research project depending on the roles and responsibilities they have. If UVM is found to be engaged in research, then our IRB must review and approve the project. See guidance below.
Review Process
Investigators may make this determination themselves, however, the IRB can acknowledge the determination.
Documentation
Any documentation generated from an acknowledgement of engagement in research is kept in a shared IRB file.
Guidance
An institution is considered engaged in human research when employees or agents for the purposes of the nonexempt research project, obtains: (1) information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or (2) obtains, uses, studies, analyzes or generates identifiable private information or identifiable biospecimens; or (3) obtains the informed consent of human subjects for the research. An institution is also considered engaged when the institution receives a direct federal award to conduct human subject research, even when all activities involving human subjects are carried out by a subcontractor.
It is not a requirement that this determination be made by the IRB, however we can review and acknowledge your determination upon request.
Category | Description of UVM Activities | UVM Engaged? | Other Site Engaged? |
|---|---|---|---|
Funding Only/No Direct UVM Involvement | UVM receives a direct grant or award to perform human subjects research, even if all of the activities are performed off-site by subcontractors or collaborators and UVM itself never intervenes or interacts directly with human subjects and never receives identifiable private information. | YES | YES |
Pre-Research Activities | UVM performs a feasibility study to determine whether sufficient data or prospective participants exist to formulate a hypothesis or conduct a study. | MAYBE | n/a |
UVM performs a small pilot study to work out details of an anticipated future research project. | YES | ||
UVM intervenes or interacts with individuals who meet study eligibility criteria to develop study protocol. | YES | ||
PR/Publicity/Pre-Screening Activities | UVM informs potentially eligible subjects about research occurring elsewhere and provides them with information about how to contact the researchers. | NO | YES |
UVM sends letters to prospective subjects describing a study and letting them know they may later be contacted to participate. | YES | MAYBE | |
Study Recruitment/Informed Consent | UVM informs prospective subjects about the availability of research conducted elsewhere; provides prospective subjects with written information about research (including the relevant informed consent document and other IRB-approved materials); provides prospective subjects with information about contacting investigators for information or enrollment; or obtains and appropriately documents prospective subjects' permission for investigators to contact them. Generally, a UVM clinician may discuss the study with the prospective subject as the subject's clinician but not as a researcher or part of the study team. *Documentation of permission must be HIPAA-compliant if the UVM investigator is affiliated with a unit covered by HIPAA. | NO | YES |
UVM faculty, staff, trainees, or agents conduct the consent interview, obtain subject signature on the consent form, or otherwise act as authoritative representatives of the investigators. | YES | YES | |
UVM researchers obtain permission from a school or nursing home to observe, audio/videotape, or distribute surveys/questionnaires for research purposes. The students or residents are consented by the UVM researchers to participate in the project. | YES | NO | |
Data/Specimen Repositories | UVM data steward queries UVM database on behalf of external researchers. | NO | YES |
UVM releases information and/or specimens to investigators at other site in non-identifiable (i.e., non-linkable) form, when the information and/or specimens were originally obtained for non-research purposes. | NO (Not Human Subjects) | NO | |
UVM receives information or specimens for research from established repositories operating in accord with an FWA, OHRP guidance on repositories, and a written agreement unequivocally prohibiting release of identifying information to UVM investigators. | NO (Not Human Subjects) | YES | |
UVM obtains, receives, or possesses identifiable (directly or with links/codes) private information from another site for research purposes. | YES | YES | |
Multi-Site Research UVM is Statistical/ Data Coordinating Center | UVM obtains, receives, or possesses identifiable (directly or through links) private information to operate a ‘statistical or data’ coordinating center for multi-site collaborative research. If the UVM investigator is within the covered entity, i.e. affiliated with a unit covered by HIPAA they are required to obtain the appropriate documentation of HIPAA compliance from the site(s) submitting identifiable private information. If UVM has no other interaction or intervention with subjects, and the principal risk associated with institutional activities is limited to the potential harm resulting from breach of confidentiality, then the UVM IRB need not review each underlying collaborative protocol. However, the IRB should determine and document that: (i) the statistical/data coordinating center has sufficient mechanisms in place to ensure the privacy of subjects and confidentiality of data are adequately maintained; (ii) each collaborating institution holds an FWA or other appropriate assurance; (iii) each protocol is reviewed and approved by the IRB at the collaborating institution prior to enrollment of subjects; and (iv) informed consent is obtained from each subject in compliance with HHS regulations, the Common Rule, and any other applicable federal policy. | YES | YES |
Multi-Site Research UVM is the Lead or Operations Coordinating Center | UVM obtains, receives, or possesses identifiable (directly or through links) private information to operate a "lead or operations coordinating center" for multi-site collaborative research. If UVM as the lead/operations coordinating center has no other interaction or intervention with subjects, the UVM IRB need not review each underlying collaborative protocol. However, the IRB should determine (i) management, data analysis, and Data Safety and Monitoring (DSM) systems are adequate, given the nature of the research involved; (ii) sample protocols and informed consents are developed and distributed to each collaborating institution; and (iii) each collaborating institution holds an applicable OHRP-approved Federalwide Assurance (FWA); (iv) each protocol is reviewed and approved by the IRB at the collaborating institution prior to enrollment of subjects; and (v) any substantive modification by the collaborating institutions of sample informed consent information related to risks or alternative procedures is appropriately justified; and (vi) informed consent is obtained from each subject in compliance with HHS regulations, the Common Rule, and any other applicable federal policy. | YES | YES |
Medical Care/ Standard Clinical Practice | UVM provides unforeseeable but medically appropriate clinical treatment incident to a patient's participation in a clinical trial elsewhere, e.g., when patient suffers an adverse event that is treated at UVM by her regular health care provider. | NO | YES |
Patient is hospitalized at UVM and attending physician decides continued participation on protocol at outside institution is ok. | NO | YES | |
Medical Care/ | UVM administers test article and performs normal monitoring, but does not perform data collection. | NO | YES |
UVM performs blood draw for a genetics study occurring at another institution but sends the samples to the other institution for analysis. | NO | YES | |
UVM provides pre- and/or post-test genetic counseling to study participants regarding tests conducted by and results reported from other institution. | NO | YES | |
Subinvestigators | UVM consents prospective subjects (even if for "someone else's study"). | YES | YES |
UVM performs physicals or other eligibility testing to be sent to investigators at another site. | |||
UVM collects and reports data to investigators at another site. | |||
Contracted Medical/ Professional Services | UVM performs physicals or other eligibility testing to be sent to investigators at another site. | NO | YES |
UVM performs and/or analyzes blood draw for genetics study occurring at other institution and furnishes the results to the investigator. | |||
UVM collects and reports data to investigators at another site. | |||
Consulting Services | Consultant does not obtain, receive or possess identifiable private information (e.g., the consultant analyzes data that cannot be linked to individual subjects, directly or through a coding system, by any member of the research team). | NO | YES |
Consultant accesses or uses identifiable private information while visiting the research team's institution. | NO | YES | |
Consultant obtains coded data for analysis at UVM (and there is not a written agreement unequivocally prohibiting release of identifying codes to the consultant). | YES | YES |
3.8 De Novo Review of Protocols
New 11/30/20
To ensure that research protocols continue to meet current regulatory and institutional standards, the IRB may require a de novo review of studies that have been modified extensively or have been open for greater than 6 years. De novo review will be at the discretion of the IRB reviewer at time of audit, modification or continuing review. The criteria for requiring a de novo review is an inability for the IRB to ensure and document the activities continue to meet current regulatory requirements thus protecting human subject rights and welfare.
De novo review requires that a new protocol submission be submitted for IRB review. Review type will be based upon risk.
4. IRB Review Determinations 46.109 and 46.113
The IRB shall review and have authority to approve, require modifications in (to secure approval), or disapprove all research activities covered by this policy, including exempt research activities under § __.104 for which limited IRB review is a condition of exemption (under § __.104(d)(2)(iii), (d)(3)(i)(C), and (d)(7), and (8)).
Approved
This decision indicates that the initial review of a research project at a convened meeting or via expedited review has been approved without requiring either further (a) changes to the protocol or informed consent document(s), or (b) submission of clarifications or additional documents.
Modifications Required for Initial Approval
This decision indicates approval is pending satisfactory resolution of conditions or clarifications that the IRB requires to approve the project. Under this scenario, for full review protocols, further review by the IRB at a subsequent convened meeting is not necessary to secure final approval.
The expedited review process is employed to review the response from the investigator. The IRB directs that the IRB chairperson (or other individual(s)) to review and determine on behalf of the IRB whether the changes, clarifications, and/or additional documents to be submitted by the investigator(s) are satisfactory.
If under the expedited review of the response, the IRB chairperson (designated reviewer) is unable to approve the project because he/she cannot make the determinations required for approval, they can either refer the project to the IRB for further review and action at a convened meeting, or defer approval of the research project and require that the investigator (a) make changes to the protocol or informed consent documents, or (b) submit clarifications or additional documents prior to further review by the IRB chairperson (or designated reviewer(s)).
Deferred
This action is taken under full Committee review any time the IRB cannot make one or more of the determinations required for approval by the HHS regulations at 45 CFR 46.111 and, if applicable, subparts B, C, or D of 45 CFR part 46, but with revisions may be found to be approvable.
When deferring a project, the IRB, under its authority to require modifications in order for an investigator to secure approval, may require that the investigator (a) make changes to the protocol or informed consent documents, or (b) submit clarifications or additional documents. The research may not proceed until the IRB reviews, at a subsequent convened meeting, the revised research project and approves it.
Disapproved
This action is taken under full Committee review when the determinations required for approval of the research cannot be made, even with substantive clarifications or modifications to the protocol and/or informed consent/process/document.
If the IRB decides to disapprove a research project, it will include in its written notification a statement of the reasons for its decision, based on specific federal regulatory criteria.
Administrative Holds, Suspensions or Terminations 46.113
All currently approved research is subject to modification or change in approval status, as deemed necessary by the IRB. The IRB has the authority to suspend or terminate research for not being conducted in accordance with State and Federal laws/regulations, and/or IRB requirements, policies and procedures; or if it has been associated with unexpected serious harm to subjects. The Investigator also has the option to place the research on administrative hold. See section Administrative Hold, Suspension, or Termination of IRB Approval.
4.1. Administrative Hold, Suspension, or Termination of IRB-Approved Approvals
Revised: 2/22/22
The IRBs have the authority to suspend or terminate their approval of research that is not being conducted in accordance with IRB requirements or federal regulations, when the research is associated with unexpected serious harm to research participants or others or when there are immediate serious issues involving participant and/or others safety. 45 CFR 46.113.
An investigator may also place a voluntary administrative hold on previously approved research when in the judgment of the investigator an administrative hold is appropriate to protect the rights or welfare of participants.
In this policy, the IRB designee refers to the following: IRB Chair, Associate IRB Chair, IRB Director, and Institutional Official.
Administrative Hold
An administrative hold is a voluntary action by an investigator to stop temporarily or permanently some or all approved research activities. Administrative holds are not considered suspensions or terminations, and do not meet reporting requirements to OHRP, FDA and other federal agencies. Administrative holds must not be used to avoid reporting deficiencies or circumstances that otherwise require reporting to federal agencies.
1. Investigator can place an administrative hold on research if; a complaint is received; an allegation of non-compliance is reported to the IRB; a discovery by the investigator of potential additional risk to subjects; or a potential change in the rights or welfare of subjects.
2. Investigator must notify the IRB in writing when exercising the option for administrative hold. The Investigator could place specific activities on hold, such as a hold on recruitment; hold on screening/enrollment; hold on interactions/interventions with subjects; and/or hold on collection or analysis of private identifiable information about subjects.
3. Investigator must address the effect of the administrative hold on the rights and welfare of the current subjects.
4. Investigator must promptly notify the IRB in writing of the intention to remove the administrative hold prior to implementing the action.
During administrative hold, the research remains subject to continuing review and requirements for reporting non-compliance and unanticipated problems involving risks to subjects or others.
The IRB designee may make recommendations for additional education and/or compliance interventions for the Investigator and research personnel. At any point, the IRB Committee can suspend the research, which will result in required regulatory reporting.
Suspension
A suspension of IRB approval is a directive of the convened IRB, or IRB designee either to stop temporarily some or all previously approved research activities, or to stop permanently some or all previously approved research activities. All suspensions are immediately reportable to OHRP, FDA and other federal agencies applicable.
Examples include an unanticipated problem in research involving greater than minimal risks to subjects or others; new information becomes available that could alter the original determination by the IRB to approve the study; (unexpected serious harm to subjects) or the PI fails or appears to fail to comply with federal regulations or UVM policy regarding the protection of human subjects.
The IRB designee has the authority to suspend previously approved research when required for the urgent protection of the rights and welfare of participants and insufficient time exists for the convened IRB to review the event. Any suspension of research by the above individuals is placed on the next available agenda, reviewed, and upheld, overturned or supplemented by the convened IRB at their meeting.
During suspension, the research remains subject to continuing review and requirements for reporting non-compliance and unanticipated problems involving risk to subjects or others.
The IRB Chair or designee notifies the investigator of the suspension and the reason for the suspension.
Safety concerns are reviewed through the Safety Subcommittee process and noncompliance issues are reviewed following the Noncompliance Policy and Procedures.
Termination
A termination of IRB approval is a directive of the convened IRB or IRB designee to permanently stop all previously approved research activities. All terminations are reportable to OHRP, FDA and other federal agencies applicable. Protocol approval will not be terminated without first undergoing temporary suspension and completion of a review through the Safety Subcommittee process or the Noncompliance Policy and Procedures.
Previously approved research may only be terminated by the convened IRB, including protocols originally approved under expedited procedures.
Terminated protocols are considered closed and no longer require continuing review.
5. Eligibility to Perform Research At UVM/UVMMC
Revised 9/13/21
Eligibility requirements for conducting human subjects research vary depending on the role of the researcher. Research personnel must be appropriately qualified by training and/or experience to perform their research responsibilities.
Principal Investigator
46.108(a)(3)(iii) “investigators will conduct the research activity in accordance with the terms of the IRB approval until any proposed changes have been reviewed and approved by the IRB…”
The Principal Investigator (PI) is ultimately responsible for assuring compliance with applicable University IRB policies and procedures, DHHS Federal Policy Regulations, FDA regulations and for the oversight of the research study, the informed consent process and the protection of individually identifiable health information. Although the PI may delegate tasks to members of their research team, the PI retains the ultimate responsibility for the conduct of the study.
Because PI responsibilities involve direct interaction and supervision of the research team, the PI must be a current employee or student at the University and/or UVMMC who is operating within their University or UVMMC role to oversee the conduct of the study. PIs leaving the institution are responsible for notifying the IRB well in advance of their departure so that they can decide to either close the study or name another appropriately qualified individual currently at the institution to serve as the PI. See manual section on Managing Research Prior to Departure.
The following individuals may serve as PI:
- Faculty members: All categories of compensated faculty members may serve as PI if their School allows them to serve as Principal Investigator on applications for sponsored funding administered through the University.
- Non-Faculty: A non-faculty researcher includes, but is not limited to, any of the following: fellow, resident, post-doctoral fellow, post-doctoral associate, post-doctoral trainee, and any student (graduate or undergraduate). Non-faculty researchers cannot conduct human subject research without having a faculty sponsor who is responsible for overseeing the conduct of the research activities. The faculty sponsor must be employed by the institution (UVM or UVM Medical Center.)
- Staff: Other University or UVMMC staff may serve in this role if they have appropriate qualifications to conduct the research and if they have obtained approval to conduct the research from their immediate supervisor.
- Students: Students may serve as principal investigators for their own research projects and are responsible for submitting the IRB application. However, when a student is listed as the PI, a faculty sponsor must be listed on the protocol submission.
Note: The IRB reviews and holds student research projects to the same standards as human subject research conducted by faculty or staff. IRB approval or exemption must be obtained prior to initiating any research activity under IRB oversight. “Retroactive” IRB approval or exemption is not permitted under federal regulations and University policy. Failure to obtain IRB approval for research with human subjects may preclude the use of the previously collected data and could result in other institutional sanctions.
Key Personnel
For expedited and full review protocols, key personnel are individuals who contribute to the scientific development or execution of a project in a substantive, measurable way, whether or not they receive salaries or compensation under the protocol. This includes, but is not limited to, individuals involved in conducting the research with human subjects through an interaction or intervention for research purposes, including participating in the consent process by either leading it or contributing to it; and those who are directly involved with recording or processing identifiable private information, including protected health information, related to those subjects for the purpose of conducting the research study.
Note: Because minimal risk studies (exempt) are defined as those where the probability and magnitude of physical or psychological harm is that which is normally encountered in daily life, it is not required to list key personnel on these studies.
Individuals who are Not Key Personnel: An individual who will be interacting with research subjects during the course of a research study, but only in his/her regular non-research employment capacity, such as a clinic receptionist, nurse or phlebotomist, or a radiologist or radiology technician, should not be listed as Key Personnel for the study if the person will perform only genuinely non-collaborative services meriting neither professional recognition nor publication privileges and not associated with individual financial gain, and will not contribute to the design, governance and/or analysis of the study.
Key personnel must complete required training and be listed as a member of the study team.
Faculty Sponsor
All research conducted by students/trainees, including postdoctoral fellows, must include a faculty sponsor as a member of the study team. In addition to the expectation that the faculty sponsor provides active mentorship to the student during the conduct of the research, the faculty sponsor shares responsibility with the student/trainee researcher for the ethical conduct of the research and is institutionally accountable for the study.
Students/Trainees
Supervision by faculty sponsors is required for any research performed by students/trainees in any role, to ensure proper conduct of research and protection of subject rights and welfare.
5.1 Responsibilities of Principal Investigators
The PI has primary responsibility for protecting the rights and welfare of human subjects in research. The PI’s primary responsibilities includes, but is not limited to, the following:
1. Delegation of Responsibilities
PIs must personally perform or delegate to qualified co-investigators or research staff all of the necessary tasks to carry out their studies. Even when specific tasks are delegated, the PI remains ultimately responsible for proper conduct of the study and fulfillment of all associated obligations.
2. Oversight of Research Team
The PI must provide members of the research team with sufficient oversight, training and information to facilitate appropriate safety procedures and protocol adherence. In addition, the IRB must be informed if a PI is no longer able to fulfill his or her duties for any reason including, but not limited to, traveling for a prolonged period of time. See PI Managing Research prior to Departure…
3. Knowledge of Human Research Protection Standards
The PI and all key personnel (together referred to as “researchers” or the “research team”) are expected to be knowledgeable about and comply with the requirements of each of the following:
• The Common Rule (link is external) and other federal research laws and regulations;
• Applicable state law;
• The University’s Federalwide Assurance;
• Institutional policies and procedures for the protection of human subjects and reporting and managing conflicts of interest;
• The terms and conditions of any research agreements (with government or private sponsors); and
• The basic ethical principles that guide human subjects research.
Institutional policies and procedures include these IRB Operating Procedures, as well as policies and procedures maintained by the academic units to which researchers and research staff are appointed, and the policies and procedures of other research review units with relevant oversight responsibilities, such as UVMMC Billing Compliance, the Institutional Biosafety Committee, etc. See Coordination with Other Compliance Committees.
4. Evaluation of Adequacy of Resources
PIs must ensure that adequate resources (facilities, equipment, supplies, and personnel) exist to:
• Conduct the research (e.g., through internal or external funding for staff, facilities and equipment);
• Protect subjects; and
• Ensure the integrity of the research.
5. Training Requirements
Researchers must complete educational training as required by the University, the relevant IRB, and other review units prior to initiating research, and should not undertake responsibility for human subjects studies unless they understand these requirements and are willing to be held accountable for complying with the relevant standards and protecting the rights and welfare of research participants. For additional information refer to Training Requirements.
6. Conflict of Interest Disclosures
Financial conflicts of interest relating to human subjects research must be disclosed. Such conflicts of interest are not inherently wrong, and as long as they are disclosed and appropriately managed or resolved, they do not distort and can benefit the research process. A conflict of interest may arise when a faculty or staff member has a relationship with an outside organization that puts the faculty or staff member in a position to influence the university's decisions in ways that could lead directly or indirectly to financial gain for the faculty or staff member or his or her family, or give improper advantage to others to the detriment of the University.
7. ClinicalTrials.gov Registration
Certain clinical studies involving human subjects must be registered on and have results posted in ClinicalTrials.gov. Please review the link below to find step by step instructions on registering your study.
https://commons.med.uvm.edu/dean/comclntril/SitePages/Registering%20a%20Clinical%20Trial.aspx
8. Studies Regulated By the Food and Drug Administration
When conducting research involving FDA-regulated products, researchers must comply with all applicable FDA regulations and fulfill all investigator responsibilities or all sponsor-investigator responsibilities, as applicable.
5.2 Key Personnel Responsibilities
This is a general guide and does not contain a comprehensive description of all of the potential responsibilities of a member of the research team (key personnel).
1. Minimizing Risks to Subjects and Protecting Subject Rights and Welfare
Federal regulations, institutional policy, and guiding ethical standards require that human subjects research be designed to minimize risks to participants. Minimizing risks and protecting human research subjects take precedence over the goals and other requirements of any research endeavor.
2. Obtaining and Documenting Informed Consent
Informed consent must be obtained from and documented for each prospective research subject (or the subject’s legally authorized representative) before the subject begins to participate in the research (including any related eligibility testing not conducted solely for clinical purposes), unless the appropriate IRB has approved a waiver or alteration of consent, or waiver of documentation.
Informed consent is not a single event or document, but an ongoing process that takes place between the investigator (or other key personnel, as appropriate) and the research subject. Informed consent requires full disclosure of the nature of the research and the subject’s participation, adequate understanding on the part of the subject (or the subject’s legally authorized representative), and the subject’s voluntary decision to participate.
3. Compliance With IRB and Other Requirements
An IRB must review and approve all research activities that meet the definition of human subjects research before they are initiated, unless an IRB has determined that the activities are exempt from IRB oversight. For non-exempt research, the IRB is also responsible for review and approval of continuing review, progress reports, change of protocol, adverse event reporting, Other Reportable Information or Occurrence (ORIO) reporting, monitoring and record keeping. Researchers must at all times cooperate with the IRB in fulfilling its responsibilities.
4. Conflict of Interest Disclosures
Financial conflicts of interest relating to human subjects research must be disclosed. Such conflicts of interest are not inherently wrong, and as long as they are disclosed and appropriately managed or resolved, they do not distort and can benefit the research process. A conflict of interest may arise when a faculty or staff member has a relationship with an outside organization that puts the faculty or staff member in a position to influence the university's decisions in ways that could lead directly or indirectly to financial gain for the faculty or staff member or his or her family, or give improper advantage to others to the detriment of the University.
5. Studies Regulated By the Food and Drug Administration
When conducting research involving FDA-regulated products, researchers must comply with all applicable FDA regulations and fulfill all investigator responsibilities or all sponsor-investigator responsibilities, as applicable.
5.3 Access To and Retention of Research Records
Access to Research Records
The investigator must provide direct access to all research records to the IRB staff. Dependent upon the protocol sponsorship there may be others with access needs, such as study monitors, FDA, and other regulatory authorities. There may be other units internally (e.g., OCTR, UVMCC, CRC, UVM Medical Center Risk Management) with oversight responsibilities that must be granted access as well.
NOTE: Unless otherwise indicated in a protocol and/or consent form, a subject’s specific research data is generally not provided to the research subject or his/her representative.
Retention of Research Records
The regulatory mandated duration for records retention varies depending on which regulations apply to the research in question. UVM investigators need to ensure that their plan for record retention complies with the federal regulations as well as UVM Records Retention policy (PDF).
- *IRB Records means all records of communications with the IRB and all approval documents.
- *HIPAA Waiver means the record of the IRB determination of a waiver of authorization.
- **Research Records (subject study file) refers to documentation of all observations and other data pertinent to the investigation on each individual in the investigation.
- **HIPAA Authorization means either the executed consent form, separate authorization or documentation of verbal authorization.
*Captured by the IRB within the electronic submission software.
**PI’s responsibility to ensure retention requirements are met.
Research Not Regulated by the FDA
45 CFR 46.115(b) (opens in a new window) (DHHS) requires that all IRB records be retained for at least 3 years, and research records be retained for at least 3 years after completion of the research. All records must be accessible for inspection and copying by authorized representatives of HHS at reasonable times and in a reasonable manner.
UVM policy requires that the IRB and investigator maintain records for 6 years due to the extended retention period required by HIPAA (see below).
Research Regulated by the FDA (Drug and Device)
21 CFR 312.62(c): (opens in a new window) An investigator involved in the research of drugs, devices, or biologics being tested in humans for FDA approval shall retain records
- for a period of 2 years following the date a marketing application is approved for the drug for the indication for which it is being investigated; or, if no application is to be filed or if the application is not approved for such indication, until 2 years after the investigation is discontinued and FDA is notified.
- The date of last marketing approval will not be known at the time the research is completed and can be quite long. Investigators are advised to include funds for storage of the case records in their study budget. Records should be retained until there is written confirmation from the sponsor or FDA granting permission to destroy them.
UVM policy requires that the IRB and investigator maintain records for 6 years due to the extended retention period required by HIPAA (see below).
Research Which Includes Protected Health Information (HIPAA Requirements)
45 CFR 164.530(j)(1) (opens in a new window) HIPAA requires a 6 year retention period for the documents listed below.
- Written HIPAA Authorizations: If the research involves Protected Health Information (PHI), the Principal Investigator must retain the permission (i.e. the consent form or authorization) to use the PHI for 6 years beyond the expiration date of the authorization (completion of the research).
- IRB Determinations for Waiver of HIPAA: Records documenting that a request for waiver of HIPAA Authorization met all the requirements of 45 CFR 164.512(i.)(2)(ii) must be retained for 6 years from the completion of the research. This information will be archived in the IRB electronic system.
- Disclosures of PHI: An accounting of all disclosures of PHI must be retained for 6 years after the disclosure. Retaining the disclosures is a responsibility of both the PI and the hospital.
UVM policy requires that the IRB and investigator maintain records for 6 years due to the extended retention period required by HIPAA.
5.4 CITI Training Requirements
Revised 5.3.2023
The University of Vermont and UVM Medical Center have pledged a commitment to the protection of all human subjects in research and have a long-standing history in extending protections required by federal funding to all research activities. In keeping with this commitment, completion of the UVM and UVM Medical Center approved human subjects training is required for all individuals involved in the conduct of research involving human subjects, regardless of funding source.
The IRB will not release the approvals for new protocols until all individuals involved with human subjects on the project (i.e., direct contact with subjects or access to data) have completed the required training as listed below. Additionally, no new personnel will be added to the protocol until the required training is complete.
The principal investigator is also responsible for ensuring the research team has appropriate protocol specific training prior to and during the conduct of the study. Dependent upon delegated responsibilities, this training could take the form of attendance at investigator meetings, regular local research team meetings, or daily mentorship from the principal investigator.
HUMAN SUBJECTS IN RESEARCH TRAINING
Please reference the CITI Program Training page on our website for additional information about required human subjects in research training. Refreshers are required every three years.
GOOD CLINICAL PRACTICE TRAINING
Good Clinical Practices (GCP) training must be completed if working on a study that meets the NIH definition of a Clinical Trial. Please reference the CITI Program Training page on our website for additional information about GCP training. Refreshers are required every three years.
Please note that principal investigators or key personnel affiliated with the UVM Larner College of Medicine are required to complete the Good Clinical Practice Training regardless of whether the research project is a clinical trial.
RESEARCH INVOLVING PRISONERS TRAINING
The principal investigator and all key personnel listed on a protocol involving prisoners as research participants must complete the CITI training titled “Research Involving Prisoners” This is a onetime training that does not have an expiration date that requires a refresher.
WHO IS REQUIRED TO COMPLETE CITI TRAINING
Principal Investigators on all active expedited, full, exempt 2(iii), 3(i)(c) and 4(iii) protocols.
Faculty Sponsors and Key Personnel listed on active expedited, full, exempt 2(iii), 3(i)(c) and 4(iii) protocols.
No CITI training is required for expanded access or “compassionate use” treatment trials. Training will not be required for all exempt protocols except exempt 2(iii), 3(i)(c) and 4(iii).
DOCUMENTATION OF COMPLETION
Each individual has an opportunity to print a CITI completion certification. Additionally, the IRB receives a completion information from CITI and places that information on our Tutorial Completion page. The types of courses and dates of completion will be automatically entered into the UVMClick system.
TRAINING EXPIRATION
Applicable training requirements must be renewed every three years. Reminders of impending expiration are sent to the individual as the anniversary nears. If a PI’s training expires, notice of this lapse in completion will be forwarded to the Department Chair. Protocol related research activities must stop until training is complete. If the study is a treatment trial and withholding the treatment is not in the best interest of the subjects, the PI must contact the Research Protections Office immediately to determine the appropriate action.
If a Faculty Sponsor’s training expires, the student PI must find an alternate Faculty Sponsor until the training is complete. The individual has 30 days to find an alternate sponsor or his/her protocol with be removed from further committee consideration.
5.5 Guidance on Data Management in Human Subjects Research
Background
This guidance is intended to assist researchers in developing data management plans for human research data. The guidance presents relevant definitions and key concepts concerning human research data, describes the roles and responsibilities for data management, outlines the elements of a research data management plan, and describes requirements for maintaining and using human research information once projects are completed.
The protection of privacy and the confidentiality of information about research subjects is a special concern for IRBs in their review of research data management. Research subjects must have a reasonable expectation that personal information will be disclosed only with their permission or in ways that are consistent with the consent process, and in compliance with the laws and regulations. Violations of confidentiality could have serious consequences for research subjects, including potential discrimination, misuse of genetic information, loss of insurance, or loss of privacy. Even with all the appropriate state and federal laws, University and hospital policies, and requirements of IRBs all aimed at protecting the confidentiality of a research subject’s individually identifiable private information, violations of privacy can and do occur. Such violations may be inadvertent (accidental) or due to carelessness, deliberate or compelled by regulation or law.
Clearly defined and faithfully followed procedures to protect the confidentiality of human subjects can significantly reduce the possibility of violations to the confidentiality of human research data and should be part of every study design.
In addition to this guidance, researchers may need technical support from either UVM’s Enterprise Technology Services (ETS) or the College of Medicine Technology Services (COMTS) for assistance with development of an adequate research data protection protocol. Contacts are listed below.
UVM Information Security Operations Team
COM IT Information Security
University of Vermont (UVM) policies referenced in this document:
Information Security Procedures
UVM Medical Center policies:
UVMMC employees may view related policies on the UVMMC Intranet.
Please note that the information in this guidance was current when the guidance was issued Summer 2015. As technologies and social norms advance, however, the standards for managing data may change.
Definitions and Key Concepts
Anonymization
This process removes information from data that allows recognition of particular individuals. Common strategies for anonymizing data are deleting or masking personal identifiers, such as name and social security number, and suppressing or generalizing quasi-identifiers, such as date of birth and zip code. Cell size restrictions may also be applied. Typically anonymized data is not coded; it ordinarily contains no link to individually identifying information that may be available to the researcher. Information that was previously recorded or collected without any of the 18 identifiers as defined by HIPAA, and no code is assigned that would allow data to be traced to an individual.
Coding
Coding is a process in which individually identifying information is replaced with a number, letter, symbol, or combination thereof and a key linking the code to identifiers is created. The key is usually maintained separately from the coded data. Coding is one means to protect the confidentiality of research data.
The HIPAA Privacy Rule requires that a code for the re-identification of health information does not derive from or be related to identifiers for an individual and not be capable of translation to identify an individual. This requirement can eliminate the use of so-called hash codes.
Confidentiality
Confidentiality means restricting access to information that an individual has disclosed in circumstances that the individual can reasonably expect the information will not be made public. The relationship between a researcher and a study participant is ordinarily one of trust. This relationship often includes an expectation that personal information collected for research that is ordinarily regarded as private will not be divulged outside the research team without explicit permission or in ways that are inconsistent with consent to research participation.
Covered Entity
Those entities to which HIPAA Privacy Rule standards apply are called “covered entities”. They are defined as (1) health insurance plans, (2) health care clearinghouses, and (3) health care providers that electronically transmit health information in connection with medical service transactions.
Data
The Merriam-Webster Dictionary (2005) defines data as "factual information (as measurements or statistics) used as a basis for reasoning, discussion, or calculation." There are many ways to categorize data; for the purpose of this guidance, the terms as used in this document are described below.
Research data refers to collected and recorded factual information commonly accepted in scientific and scholarly communities as necessary to validate research findings. Research data can be classified as:
- Anonymous research data: Research data that lacks information that would allow the recognition of particular individuals by the researcher.
- Health data: Health information created or received by health care providers, insurance plans, and clearinghouses that is individually identifiable is protected by federal and state laws, including the Health Insurance Portability and Accountability Act of 1996 (HIPAA; see the definition below.).
- Identifiable biospecimen/identifiable private information. An identifiable biospecimen is a biospecimen for which the identity of the subject is or may readily be ascertained by the investigator or associated with the biospecimen.
- Identifiable research data: Research data containing information that allows recognition of particular individuals from the data by the researcher.
- Indirectly identifiable research data: Research data that are coded with a key linking the data to individually identifying information. The key may or may not be available to the researcher.
- Limited data set: See the definition below.
- Processed research data: Analyses, descriptions, and conclusions prepared as reports, manuscripts, theses, or papers.
- Published research data: Written information distributed to people beyond those involved in research data acquisition.
- Raw or primary research data: Information recorded as notes, images, video recordings, paper surveys, computer files, etc., pertaining to a specific research project. Examples of such data are: survey responses, observations of behaviors, observations of medical symptoms, temperature readings, behavioral or medical test results, biological samples, and radiographic images.
De-Identification
As required by the HIPAA Privacy Rule, this process involves the removal of the following informational elements from health information.
1. Names.
2. All geographic subdivisions smaller than a state, including street address, city, county, precinct, ZIP Code, and their equivalent geocodes, except for the initial three digits of a ZIP Code if, according to the current publicly available data from the Bureau of the Census:
- The geographic unit formed by combining all ZIP Codes with the same three initial digits contains more than 20,000 people, and the initial three digits of a ZIP Code for all such geographic units containing 20,000 or fewer people are changed to 000.
3. All elements of dates (except year) for dates directly related to an individual, including birth date, admission date, discharge date, date of death; and all ages over 89 and all elements of dates (including year) indicative of such age, except that such ages and elements may be aggregated into a single category of age 90 or older. The population of a zip code can be identified on the web site of the U.S. Census Bureau at the following url: http://www.census.gov/popfinder/
4. Telephone numbers.
5. Facsimile numbers.
6. Electronic mail addresses.
7. Social security numbers.
8. Medical record numbers.
9. Health insurance plan beneficiary numbers.
10. Account numbers.
11. Certificate/license numbers.
12. Vehicle identifiers and serial numbers, including license plate numbers.
13. Device identifiers and serial numbers.
14. Web universal resource locators (URLs).
15. Internet protocol (IP) address numbers.
16. Biometric identifiers, including fingerprints and voiceprints.
17. Full-face photographic images and any comparable images.
18. Any other unique identifying number, characteristic, or code, unless permitted by the HIPAA Privacy Rule standard for re-identification.
In addition, health information can be de-identified if (a) a person with appropriate knowledge of and experience with generally accepted statistical and scientific principles and methods determines that the risk is very small that the health information could be used to identify an individual and documents the methods and results of this analysis, or (b) the covered entity from which health information is being obtained does not have actual knowledge that the information could be used to identify an individual.
Encryption
Encryption is the process of comprehensively encoding information in such a way that only authorized parties can read it. Encryption limits access to data by controlling the distribution of the decoding algorithm to authorized individuals. Encryption is one means of protecting the confidentiality of research data. Under HIPAA, if protected health information is encrypted, this action provides a “safe harbor” from violating HIPAA.
Health Insurance and Portability and Accountability Act of 1996 (HIPAA)
HIPAA is federal legislation that, along with its implementing regulations, produced legal protections for health information. This legislation has had an impact on the conduct of research involving health information. A critical part of HIPAA for research activities is the so-called privacy regulations, often referred to as the Privacy Rule. The intent of the HIPAA Privacy Rule is to protect the confidentiality of health care information and define the rights of patients regarding their health information. The HIPAA Privacy Rule created the concept of “protected health information” or “PHI”, which is individually identifiable health information created or received by a covered entity (See the definition of covered entity above.)
Under the HIPAA Privacy Rule, specific permission from patients for research use of health information may be obtained using an Authorization in addition to the consent process. The HIPAA Privacy Rule specifies the kinds of information included in the Authorization language. Provided certain regulatory criteria are satisfied, an IRB may waive or alter the information included in the Authorization language.
Honest Broker
An honest broker obtains legally protected data from their source and typically codes and then de-identifies the data or creates a limited data set for research use. The honest broker retains the key linking the code to identifiers for individual contributors of the data. If questions arise about the validity or accuracy of the data, the honest broker can typically resolve them without revealing the identity of individuals to the researcher. As a result of the activities of the honest broker, the research conducted with processed data may not involve human subjects, because the data lacks identifiable private information.
An investigator actively involved in the research in which the data are used cannot be an honest broker, nor can any person under the investigator’s supervision.
Identifiers
Identifiers are specific informational elements that permit the recognition of a particular person.
Limited Data Set
Under the HIPAA Privacy Rule, a limited data set is defined as protected health information that excludes the aforementioned direct identifiers of an individual or of relatives, employers, or household members of that individual.
A limited data set may only be used for the purposes of research, public health, or health care operations.
A data use agreement between the source of the protected health information and the recipient is needed for use of a limited data set. This agreement restricts the uses and disclosures of the limited data set, requires the recipient to establish appropriate safeguards to limit further uses and disclosures, applies the HIPAA Privacy Rule to the use of the limited data set, and prohibits identifying or contact with the individuals who contributed the data.
Repository
A repository compiles data, specimens, or both for future research purposes. The repository receives, processes, stores, and distributes data with or without specimens to researchers. The repository may or may not be an honest broker.
Sensitive Data
Data that are individually identifiable and private usually are considered sensitive and should be protected appropriately. Identifiable data that if disclosed could have adverse consequences for subjects or could damage their financial standing, employability, insurability, or reputation are considered highly sensitive. The IRB may recommend that a Certificate of Confidentiality be applied in these cases to allow researchers to refuse to disclose names or other identifying characteristics of research subjects in response to legal demands. A list of research in which resultant data would be considered highly sensitive can be found in Appendix M., Section 1. Very stringent security precautions need to be in place to protect research data while in storage or being transferred.
Roles and Responsibilities for Human Subject Data Management
Principal Investigators (PIs)
PIs are responsible for developing an appropriate data management plan as well as ensuring that research staff members are thoroughly trained to maintain the integrity of the research data that is collected. These responsibilities include determining how to best collect, store, protect, analyze, and disseminate research data.
Research Team Members
The primary research data management responsibilities of research coordinators, research assistants, and those in similar positions are usually involved with research data collection, ensuring the reliable and accurate collection of the research data and protecting the confidentiality of research data that are collected.
Statisticians are primarily responsible for ensuring comprehensive and appropriate research data analysis.
The Institutional Review Boards (IRBs)
The IRBs are responsible for review and approval of proposed data management plans so that the rights and welfare of research subjects are acceptably protected.
The Research Data Management Plan
A summary of four basic types of research data appears in the table below.
DIRECTLY IDENTIFIABLE RESEARCH DATA | INDIRECTLY IDENTIFIABLE RESEARCH DATA | ANONYMIZED/RESEARCH DATA | DE-IDENTIFIED RESEARCH DATA |
|---|---|---|---|
Data contains informational elements that allow the data to be associated with a living unique individual. | Direct identifiers among the data are replaced by a code and a key to the code links it to individual identities. Re-identification of the data is possible. | Individual identifiers were never recorded or have been stripped from the dataset and the data has been manipulated to make it very difficult to re-identify individuals. The data is not coded. | Certain specified informational elements are absent from the data. Re-identification of the data is not possible. |
Before starting a new scientific research project, the PI and research team should address the following activities related to research data management.
Data Collection
The information below is intended as general advice to researchers about devising a data management plan.
The data management plan should reflect whether or not the project requires that data to include
- Direct identifiers,
- If a code should be used, resulting in indirectly identifiable data,
- If the research data should be anonymized, or
- If the data should be collected without any identifiers at all.
The use of identifiers should have a clear justification, because it will increase risks to the confidentiality of subject data. If the research data is coded, the plan should describe the coding methodology, and the security arrangements for storage of the “key” linking the code to identifiers. In general, the key should be stored separately from the data.
Data Storage
Researchers should decide how they are going to store research data, in what format, and for how long. Considerations should include the data retention requirements of UVM and UVMMC, sponsors, and publishers, plus intellectual property protection, and potential data sharing.
Generally speaking, enough research data should be retained so that the findings of a project can be reconstructed with ease. While this does not mean that a project needs to retain all the raw research data that were collected, relevant statistics and analyses from this research data should be saved, along with any notes or observations.
Some key issues for electronic research data storage are: (1) thorough documentation to allow research data to be appropriately used in the future and (2) using storage formats that are adaptable to evolving computer hardware and software. There are also some additional considerations that are unique to electronic research data storage, including rapid access to the data, fast read/write rates, ability to archive and remove the data, low cost, and a backup system.
Data Retention and Disposal
Research sponsor requirements, clinical trial contracts, federal grant terms and conditions, data sharing plans, intellectual property protection, publishers’ policies, and the potential future value of the research data often require long retention periods. In addition, UVMMC’s policies apply to clinical care and other health services delivery data. In general, the UVM records retention policy applies. It can be found at http://www.uvm.edu/policies/general_html/recordretention.pdf
The IRB strongly recommends that researchers remove direct identifiers, such as those listed in the HIPAA Privacy Rule standard for de-identification, so that the identity of individual research subjects cannot be readily ascertained from the data. Additionally, researchers should arrange to securely archive signed consent forms. Such procedures for stored research data serve to minimize risks to subjects. The IRB review process includes asking researchers to describe their plans and procedures for long-term maintenance of research data involving human subjects when study protocols are closed.
Destruction of Research Data
When researchers decide that research data should no longer be maintained, the data should be thoroughly and completely destroyed. Effective destruction ensures that research data cannot be extracted or reconstructed. Many document storage companies now offer onsite shredding and secure destruction of written and electronic media.
For electronic research data, the IRB advises researchers to contact either the College of Medicine Technology Services COMTS or Enterprise Technology Services (ETS) to assist with development of an adequate data destruction plan, as simply deleting the data files is insufficient.
Data Security
Research data management plans should ensure that hard copy and electronic research data are securely stored to prevent unauthorized access, disclosure, or loss. Hard copy records should be stored in a manner that limits access to authorized individuals. For example, filing cabinets/areas should be locked and placed in rooms that are routinely locked when not in use.
Electronic research data should be stored on a device that has appropriate security safeguards, such as unique identification of authorized users, password protection, encryption, automated operating system patch (bug fix) management, anti-virus controls, firewall configuration, and scheduled and automatic backups to protect against research data loss or theft. If a researcher chooses to store directly identifiable private research data locally on the computer’s hard drive, that computer, whether a laptop or desktop, must be encrypted. The use of the network and servers maintained by the University is preferable to saving data on a local hard drive.
Laptops, Smart Phones, Tablets, removable hard drives, “jump” or “thumb” or “flash” drives, CDs, DVDs, and other portable devices and removable media are convenient to ensure your research data are always at your fingertips. External hard drives are a cost effective and convenient way to back up your research data. These devices, however, require encryption solutions if they are used to store or transfer directly identifiable private information. This requirement may be waived depending upon the sensitivity of the data being collected. The IRB will make that determination.
Because email is not secure, directly identifiable private research data that will be transferred via email, requires that the data file be encrypted prior to sending.
The IRB advises researchers to refer to the University Information Security Procedures Policy or to contact the appropriate technical support from either the College of Medicine Technology Services (COMTS) or Enterprise Technology Services (ETS) for assistance with development of an adequate research data protection protocol.
Data Analysis and IRB Approval
Primary Analysis
Primary analysis refers to how raw research data are chosen, evaluated, and interpreted into meaningful and significant conclusions. Once the primary research data analysis for a protocol is complete, the protocol should be closed with the IRB. Any subsequent analysis of the same research data, however, is usually considered secondary research data analysis.
During analysis, when subjects are typically no longer being enrolled, a researcher may apply for IRB approval to anonymize the research data by completing a continuing review application describing how identifiers will be removed. When the IRB approves a proposal to remove identifiers from remaining research data, IRB oversight of the study stops, and the researcher may continue to analyze the now anonymized research data.
Secondary Analysis
Any new, subsequent secondary analysis of existing human subject research data either requires: 1) IRB review and approval prior to access to or access to the research data; or 2) an IRB determination that its review is not necessary. See the chart below.
The IRB is responsible for determining whether or not 1) secondary analysis of research data increases risks to subjects, and 2) subjects were adequately informed during the original consent process about the possibility of secondary research use, maintenance of confidentiality, and destruction of identifiers. Based on these determinations the IRB may require the investigator to obtain informed consent from the subjects for secondary analysis.
Unless a dataset is anonymous, namely contains no direct identifiers and no code linked to identifiers, investigators who obtain research data from other researchers for secondary analysis should obtain IRB review and approval, or the IRB’s determination that the research does not require further IRB review, prior to obtaining the research data. The researcher providing the data may need to consult with his or her institution before sharing research data with local investigators. Guidance about data acquisition, management, sharing, and ownership at UVM can be found at the following URL. http://www.uvm.edu/spa/?Page=dataacquisition.html
IRB REVIEW OF ADDITIONAL DATA ANALYSIS
AFTER INITIAL PROTOCOL APPROVAL
Does The Research Data Have Identifiers? | Proposed New Analysis Plan | Requirement For IRB Review |
|---|---|---|
No | Change to analysis of anonymous or anonymized data in an existing research project | No |
Yes | Change to data analysis for a currently approved research protocol. | Submit a modification to original study for IRB review of the proposed modifications Likely to qualify for Expedited Review unless risks to subjects would be increased. |
| Yes | Analysis of research data from previously approved research after the original study is closed. | Submit a new application for IRB review with a protocol describing the research data and its analysis plan. Include a copy of the IRB approval letter and IRB approved consent form for the original study that collected the data. Note: If the original IRB approved protocol and consent did not include information about the new proposed analysis, the application for review should include a procedure for obtaining consent or a request for waiver of informed consent and authorization if PHI is involved Likely to qualify for Expedited Review unless risks to subjects would be increased. |
| Yes | Storage of research data to share with colleagues or students in the future. | Submit a new application for review of a repository. |
5.6 Managing Research Prior to Departure, Sabbatical, Medical Leave, or Other Absence
RPO requires that a qualified PI be assigned to each protocol at all times. All investigators have the responsibility to manage the disposition of their studies before taking a leave or departing the University. Department chairs or faculty mentors are responsible to ensure that this be accomplished prior to the investigator’s leave.
Procedures for a Planned Temporary Leave of Four Weeks or More
Examples of temporary leave includes, but is not limited to: Military Leave, Extended Sick Leave
In these situations, the PI must contact the IRB Research Analyst assigned to your department to discuss the specific status of the protocol(s) to assist you in making a determination as to how best to ensure continued research compliance during the leave.
Procedures for an Unforeseen Temporary Leave of Four Weeks or More
- The PI or their representative, such as the Department Chair or key research personnel, must immediately contact the IRB with the name of a designated investigator.
- The PI or their representative must submit a modificationto designate an interim investigator.
- The PI or their representative is responsible for quickly orienting key research personnel and the designee.
Procedures for Permanent Leave
Examples of permanent leave includes, but is not limited to: Graduating, Transferring to another school, indefinite break from studies at UVM, Resignation, Retirement
If intent is to close protocol
- The PI is responsible for submitting a final report in advance of IRB approval expiration.
- Student PIs who are graduating must submit a final report.
- In both of these cases, the PI can meet this requirement by completing final continuing review form.
If intent is to transfer to another UVMMC/UVM PI
- The PI must submit a modification to appoint the new PI well in advance of their departure. PIs are encouraged to seek the assistance of their Department Chair and colleagues in this process.
- If a faculty sponsor wishes to continue a student-led project after the student has graduated, they are responsible to identify a new PI or become the PI of record for the protocol. A modification to update this information must occur in advance of the student’s departure.
- In both of these cases, all protocol documents that contain the departing PI, such as the protocol, informed consent, parental permission, assent, advertisement/s, and/or recruitment material/s must be revised and submitted for approval.
Informing Subjects
If the departing PI will no longer be listed on a clinical study that includes a drug, device, or other intervention, active subjects (and any subject with an implantable device) should must be notified and provided with new contact information should they have questions about the research.
Key Personnel Who Remain Listed on a Protocol After They Leave
Key personnel who have left the institution must be removed from the protocol roster. If key personnel leaving the institution wish to continue to be engaged in the UVM/UVMMC research protocol, the protocol must be modified to reflect that change and they must obtain IRB review and approval of the protocol from their new institution prior to beginning activities. If their new institution does not have an IRB, the PI should contact the RPO office to discuss options early in the process.
Nine-Month Faculty Appointments
Nine-Month faculty are expected to be available during the summer months. If the PI anticipates not being available by any means, the PI must submit a modification identifying a person who will oversee the research during that time.
Retired Faculty with Emeriti Status
The bestowal of Emerita/Emeritus status is an honor awarded to eligible individuals who have achieved a career of professional accomplishment and provided distinguished service to the University, the community, and their professional discipline or profession.
See the University’s Emeriti Status policy. https://www.uvm.edu/policies/general_html/emeritastatus.pdf
Emeriti faculty may continue to conduct research activities if they have been provided with appropriate resources and there is adequate oversight by the academic department. The IRB may request confirmation of approval from the Department Chair prior to approving research proposed by the emeriti faculty.
Additional Things to Consider When Departing the Institution
Disposition of the Research Data
PIs are responsible for data collection, maintenance, and retention of University-owned Research Data in accordance with the Access and Retention of Research Data policy http://www.uvm.edu/spa/?Page=dataacquisition.html.
Disposition of Biological Materials
If a researcher wishes to take biological materials collected at the University of Vermont/UVMMC to continue his/her research at another institution, the consent of the Department Chair and IRB shall first be obtained. Transfer requests are subject to all terms of funding agreements under which the tissue was collected or the bank was established. The human biological materials must be transferred pursuant to a University Material Transfer Agreement ("MTA") executed by the Office of Technology Commercialization.
5.7 Data and Biospecimen Sharing
New Research Project
Researchers who intend to share research data or biospecimens with colleagues should be sure to include the intention to share materials within the initial protocol submission to the IRB. The IRB strongly recommends that direct identifiers be removed from the data or biospecimens prior to release; if UVM researchers intend to maintain identifiers, even if indirectly through use of a code, there should be an appropriate scientific justification for doing so included in the protocol. If there is written consent, the consent should include information about the details of what is being shared with whom and how it will be protected if directly identifiable.
Ongoing Research Project
If researchers find the need, or wish to share research data or biospecimens after the study has been IRB approved, the protocol must be amended prior to release. If a written consent exists, subjects should be re-consented to now share their data or biospecimens.
Original Research Project Closed
Research data sharing may occur after the original study has been completed. If a researcher intends to share research data or biospecimens from a study that has been closed, then the researcher should consult with the IRB. The consultation with the IRB is needed to determine whether or not the consent process for the original study included sharing. Dependent upon the consent process, data and/or biospecimens may need to be completely de-identified prior to release. The IRB will work with you on the available options.
Agreements to Address Sharing
In all cases above, UVM investigators sharing research data or biospecimens outside of the institution must enter into contractual agreements with the researchers to whom they are sending materials. If you are sending data, contact Sponsored Project Administration for further information regarding negotiation of a Data Use Agreement (DUA). If you are sharing biospecimens, contact the UVM Office of Technology Commercialization to determine if a Materials Transfer Agreement (MTA) or other agreement is needed. See Collaborative Agreements section.
5.8 Enrollment Incentives
Enrollment incentives are any form of direct or indirect inducement (cash or non-cash) offered or received in exchange for enrolling subjects into research studies: (i) that is paid as reimbursement in excess of the reasonable cost of conducting the research protocol, or (ii) that is paid extra-contractually, including any bonus, reward, award, grant, gift, benefit, or other quid pro quo, or (iii) that creates a financial incentive that UVM and/or UVM Medical Center determine is contrary to the best interests of human subjects participating in the research.
The use of enrollment incentives in research involving human subjects creates a significant potential for conflicts of interest. Enrollment incentives are not reasonable payments made to subjects for their participation in research or to the actual costs researchers incur when enrolling subjects. Enrollment incentives are any special incentives, finders fees, bonuses or other similar forms of compensation provided to institutions or researchers as a means to encourage the enrollment of subjects in research, including clinical trials. Such incentives may create conflicts of interest. Enrollment incentives may also have an adverse effect on human subjects because such incentives may compromise the informed consent process or increase the likelihood for enrollment of ineligible persons as participants in the research.
It is the policy of the Committees on Human Research not to approve human subjects research involving use of enrollment incentives. This prohibits any payment to investigators, study personnel or departments for additional compensation that: 1) encourages the recruitment of subjects (whether it is for identification, referral, recruitment, or enrollment of any subject); or 2) is tied in any way to the rate or timeframe for recruitment or enrollment.
6. Conflict of Interest
Revised: 08/03/2022
A conflict of interest arises with respect to activities that compromise, or appear to compromise, an employee’s judgement in performing his or her duties. These conflicts can arise when an employee, or a member of his/her family has an existing or potential personal, financial, or other interest that: (a) impairs or may reasonably appear to impair his/her independence of judgement in the discharge of responsibilities to a research participant; or (b) may result in personal gain or advancement at the expense of a research participant.
Conflicts of interest must be: (1) disclosed, (2) eliminated or (3) properly managed.
Investigators with Sponsored Research Disclosure Requirement
The Research Integrity Office has a separate policy “Financial Conflict of Interest in Sponsored Research” for sponsored research that applies to all investigators defined as the Principal Investigator (PI), Project Director (PD) or any other person (key personnel), regardless of title or position, who is responsible for the design, conduct, or reporting of research proposed to, funded by, external sponsors, under grants, contracts, cooperative agreements, or other awards for research. The same UVMClick on-line system is used for these disclosures. Disclosures are required at the following times:
(1) No later than at time of a proposal submission for externally sponsored research;
(2) At least annually thereafter during the period of research activity;
(3) During the research within thirty days of the discovery or acquisition of a new reportable financial interest; and
(4) At the time of award for Investigators whose work is funded by a sponsor that follows the Public Health Service (PHS) or the National Science Foundation FCOI Policies.
An IRB Committee member serves on the Conflict of Interest Committee to provide protocol information as needed.
Investigators with Sponsored and Non-Sponsored Research Disclosure Requirement
As part of the protocol submission, investigators must inform the Institutional Review Board (IRB) whether they or key personnel on the protocol have a significant financial interest as defined in this policy, and must describe the nature of that interest.
Institutional Review Board Committee Members Disclosure Requirement
New member initial training includes a discussion of real or perceived conflicts in relation to Committee work and review of protocols. IRB leadership, staff and affiliated IRB members disclose significant financial interests (SFI) in accordance with the UVM Conflict of Interest and Commitment and Financial Conflict of Interest in Sponsored Research policies.
Selection of specific protocols for review by members is determined by the Committee's administrative staff and/or Chair. If an IRB member has a conflicting interest in a protocol (including, but not limited to being a principal investigator, a co-investigator, or a consultant on that protocol), that member may only provide information as requested by the IRB and will not be assigned to officially review nor vote on that protocol. Members must recuse themselves from committee discussion and cannot count toward a quorum with respect to that protocol.
Definition of a Significant Financial Interest
The IRB uses the Sponsored Project Administration regulatory definition of significant financial interest for sponsored research. Significant financial interest means a financial interest consisting of one or more of the following interests of the Investigator, the Investigator’s spouse and/or the Investigator’s dependent children that reasonably appears to be related to the Investigator’s Institutional Responsibilities:
- With regard to any publicly traded entity, a Significant Financial Interest exists if the value of any remuneration received from the entity in the twelve months preceding the disclosure and the value of any equity interest in the entity as of the date of disclosure, when aggregated, exceeds $5,000.
For purposes of this definition, remuneration includes salary and any payment for services not otherwise identified as salary (e.g., consulting fees, honoraria, paid authorship), and equity interest includes any stock, stock option, or other ownership interest, as determined through reference to public prices or other reasonable measures of fair market value; - With regard to any non-publicly traded entity, a Significant Financial Interest exists if the value of any remuneration received from the entity in the twelve months preceding the disclosure, when aggregated, exceeds $5,000, or if the Investigator (and/or the Investigator’s spouse or dependent children) holds any equity interest (e.g., stock, stock option, or other ownership interest); or
- Intellectual property rights and interests (e.g., patents, copyrights), upon receipt of income related to those rights and interests.
- For PHS funded research only, any travel expenses reimbursed directly to an Investigator or paid directly on the Investigator’s behalf, regardless of amount or value, related to his/her Institutional Responsibilities, excluding travel that is reimbursed or paid by a federal, state, or local government agency, an institution of higher education (including the University of Vermont), an academic teaching hospital (including the University of Vermont Medical Center), a medical center, or a research institute that is affiliated with an institution of higher education
IRB Minimization of Risk
The IRB will evaluate all disclosures of significant financial interests, whether the disclosure is reported by the FCOI Committee or reported by the investigator through the UVM-Click IRB system. A conflict can occur with the Principal Investigator or any key personnel listed on the protocol.
The IRB shall determine whether the disclosed interests could potentially compromise or influence the investigator’s professional judgment or actions in the performance of the study or could otherwise adversely affect the rights and welfare of human participants.
The IRB will carefully consider protocol-specific mechanisms proposed by the investigator to minimize the potential adverse consequences of the conflict. For sponsored research, existing management plans as approved by the FCOI Committee, will be taken into consideration. In general, the investigator should not be directly engaged in those aspects of the trial that could be influenced inappropriately by that conflict. These could include: the design of the trial, monitoring the trial, obtaining the informed consent, adverse event, and unanticipated problem reporting, and analyzing the data. In all cases, good judgment, openness of process, and reliance upon objective, third party oversight may effectively minimize the potential for harm to participants and safeguard the integrity of the research.
During the study, new information regarding the conflict must be disclosed to the IRB in a timely manner.
Disclosure to Potential Participants
As part of the informed consent process, potential participants will be informed of the existence of relevant significant financial interests (as defined above) held by the investigator(s). Language can be found in the consent template on the IRB forms page.
7. Non-Faculty Researcher Requirement
Definition
A non-faculty researcher includes, but is not limited to, any of the following: fellow, resident, post-doctoral fellow, post-doctoral associate, post-doctoral trainee, and any student (graduate or undergraduate).
Non-faculty researchers cannot conduct human subject research without having a faculty sponsor who is responsible for overseeing the conduct of the research activities. The faculty sponsor must be employed by the institution (UVM or UVM Medical Center.)
Role of the Faculty Sponsor
The faculty sponsor will assume the role of the responsible investigator on all research involving human subjects which is designed and carried out by non-faculty. The responsible investigator will advise the non-faculty researcher throughout the process of protocol development, submission, and review, as well as in the implementation of the research project. As the responsible investigator, the faculty sponsor or course instructor is required to complete the required human subjects training. Protocol approvals will not be released until that requirement has been met.
The faculty sponsor, as the responsible investigator, will guide the non-faculty researcher in the development of the protocol, thus assuring that the content, quality, and timing of the submission meet the requirements of the Committee.
Furthermore, the faculty sponsor is accountable for ensuring that non-faculty researchers are aware of their responsibilities as investigators, and for ensuring that the Committee is immediately notified in the event of research-related, unanticipated events or findings during the study that would affect the risks or benefits of participation.
Role of the Non-Faculty Researcher
Non-faculty researchers have responsibilities as listed in “Investigator Responsibilities.”
7.1 Student Class Project Guidelines
Deciding Whether IRB Review is Necessary
The majority of classroom projects will not be considered research by Federal definition and will not require IRB review. If the intention of the class project meets the following criteria, then, the project would not meet the definition of research and does not require IRB submission.
- It is an activity designed as part of a course requirement for purposes of learning research methods and;
- The results and data will not be presented a research findings in any presentation, conference, publication, thesis, dissertation, or report outside of the course for which it is assigned.
If the intention is to present the outcome as research outside of the classroom, the project requires review and approval or a determination of exemption by the IRB prior to the start of project activities. As new researchers, student projects should fall into the exempt or expedited categories. Thus, instructors should encourage students to develop minimal risk protocols. The higher the risk, the stricter the regulations, which adds complexity for which the student is not prepared and which often times results in extended delays in IRB approval. Regardless of risk, the IRB process should be begin as soon as possible. Note that the IRB does not have the option of granting “retroactive” approval after research is complete; consult with the Research Protections Office for guidance prior to conducting research if there is any question.
Instructors should reference the IRB Review guidance for information regarding different levels of risk and types of IRB Review.
Examples Not Requiring IRB Review
- CLASS PROJECTS involving secondary data analyses that are assigned and conducted as educational exercises, using data that are either publicly available data, de-identified or otherwise impossible to be linked to personal identities.
- CLASS PROJECTS involving secondary data analyses that are assigned and conducted as educational exercises, and that use datasets that include private information and codes that link to identifiers, but the students do not have access to the identifiers.
- CLASS PROJECTS or PRACTICA that involve direct interaction (e.g., in person, via mail, email, web surveys, or telephone), but where the purpose is training, an educational exercise or professional development, and not considered research by Federal definition. The project or practicum is not “research” even if students ask people questions as part of learning how to conduct interviews or surveys, take histories, administer assessments, or perform “in-house” evaluations as requested by the practicum site.
Examples Requiring IRB Review
- If a student decides after the completion of a practicum activity to pursue additional activities with the same information for a master’s project or paper, then an IRB application describing research use of secondary data should be submitted for approval, as above.
- CLASS PROJECTS or PRACTICA that involve direct interaction or secondary analyses of private identifiable data and are undertaken as both an educational experience and as research (e.g., results of these activities will be presented publicly or otherwise disseminated, or the data will be stored and used by the students or others as research data).
Responsibility of Course Instructors
The instructor/faculty must complete the required CITI human subjects in research training.
To ensure ethical conduct of student class projects, instructors who assign a class project are expected to review student plans prior to subject recruitment and data collection.
When a student project does require IRB review, the Instructor must comply with the responsibilities as listed under Investigator Responsibilities, as well as additional mentoring responsibilities as listed below:
1. Review each student project prior to submission to the IRB for accuracy and completeness.
2. Assist and support the student in his/her interaction with the IRB and oversee resolution of issues that arise during the review process.
3. Oversee of the student’s research to ensure that human subjects are protected, e.g., the protocol is followed as approved, any unanticipated events are reported as required, etc.
4. Verify that prior approval of Thesis or Dissertation Committee, if applicable, has been obtained.
Responsibility of Students
Student researchers have responsibilities as listed under Investigator Responsibilities.
8. Types of Research
| Types of Research | Notes |
|---|---|
| Applied | Scientific investigations conducted to answer specific clinical questions or solve practice-related problems. |
| Basic | Scientific investigation that involves the generation of new knowledge or development of new theories; its results often cannot be applied directly to specific clinical situations. |
| Correlational | The systematic investigation of relationships among two or more variables, without necessarily determining cause and effect. |
| Descriptive | Research that provides an accurate portrayal of characteristics of a particular individual, situation, or group. These studies are a means of discovering new meaning, describing what exists, determining the frequency with which something occurs, and categorizing information. |
| Ethnographic | The investigation of a culture through an in-depth study of the members of the culture; it involves the systematic collection, description, and analysis of data for development of theories of cultural behavior. |
| Experimental | Objective, systematic, controlled investigation for the purpose of predicting and controlling phenomena and examine probability and causality among selected variables. |
| Exploratory | Studies that are merely formative, for the purpose of gaining new insights, discovering new ideas, and increasing knowledge of phenomena. |
| Grounded Theory | A research approach designed to discover what problems exist in a given social environment and how the persons involved handle them; it involves formulation, testing, and reformulation of propositions until a theory is developed. |
| Historical | Research involving analysis of events that occurred in the remote or recent past. |
| Phenomenological | An inductive, description research approach developed from phenomenological philosophy; its aim is to describe an experience as it is actually lived by the person. |
| Qualitative | Research dealing with phenomena that are difficult or impossible to quantify mathematically, such as beliefs, meanings, attributes and symbols. |
| Quantitative | Research involving formal, objective information about the world, with mathematical quantification; it can be used to describe test relationships and to examine cause and effect relationships. |
Note: The practice of telephone screening and consent for potential subjects is considered research and could occur in any of the above types of research.
8.1 Standard Clinical Trial Protocol
A complete protocol is required for Committee review. Researchers who are participating in a multi-center protocol may submit the lead investigator/sponsor’s protocol. The Committee requires that the “Human Research Protocol” form be utilized any time a local researcher is writing his/her own protocol, or when a grant is being submitted for review. This form includes all of the elements as listed below in a format that is easy to complete and easy for Committee members to review. The form can be found on our forms page.
The “Qualitative Research Protocol Form” should be used instead of the “Human Research Protocol” form when submitting qualitative research or primarily qualitative research and when medical procedures are not included in your research. See the section on Qualitative Research Protocol for more information.
8.1.1 Elements Found in a Standard Protocol
Revised 10/02/20
The UVMClick software will drive you through the questions necessary to ensure the required elements are present in your submission.
Introduction
The importance of the research and the potential knowledge to be gained should be explained in detail. Give background information, including references to prior human or animal research and references that are relevant to the design and conduct of the study.
Objectives
Clearly state the primary objective(s) of the study.
Study Design
Describe the research design and the procedures to be used to accomplish the specific aims of the project. Include how the data will be collected, analyzed, and interpreted as well as the data sharing plan as appropriate. Describe any new methodology and its advantage over existing methodologies. Discuss the potential difficulties and limitations of the proposed procedures and alternative approaches to achieve the objectives. As part of this section, provide a tentative sequence or timetable for the project. Point out any procedures, situations, or materials that may be hazardous to personnel and the precautions to be exercised.
Inclusion/Exclusion Criteria
Eligibility and ineligibility criteria should be specific. Changes to the eligibility criteria at a later phase of the research have the potential to invalidate the research.
Methods
Describe the types, frequency and duration of tests, study visits, interviews, questionnaires, etc. Include required screening procedures performed before enrollment and while on study.
Withdrawal Procedures
Define the precise criteria for withdrawing subjects from the study. Also include a description of study requirements for when a subject withdraws him or herself from the study (if applicable).
Statistical Considerations
Delineate the precise outcomes to be measured and analyzed. Describe how these results will be measured and statistically analyzed. Delineate methods used to estimate the required number of subjects. Describe power calculations if the study involves comparisons. Perform this analysis on each of the primary and secondary endpoints, if possible.
Human Subject Protections:
Subject Selection: Provide rationale for subject selection in terms of the scientific objectives and proposed study design.
Include as appropriate:
Inclusion of Minorities and Women: Describe efforts to include minorities and women. If either minorities or women are excluded, include a justification for the exclusion.
For protocols including the use of an investigational drug, indicate whether women of childbearing potential have been included and, if not, include appropriate justification.
If HIV testing is included specifically for research purposes explain how the test results will be protected against unauthorized disclosure. Include if the subjects are to be informed of the test results. If yes, include the process and provision for counseling. If no, a rationale for not informing the subjects should be included.
Special Populations: Explain the rationale for involvement of special classes of subjects, if any. Discuss what procedures or practices will be used in the protocol to minimize their susceptibility to undue influences and unnecessary risk (physical, psychological, etc.). See section: Additional Protections for Special Populations
Inclusion of Children: Describe efforts to include children. Inclusion is required unless a clear and compelling rationale shows that inclusion is inappropriate with respect to the health of the subjects or that inclusion is inappropriate for the purpose of the study. If children are included, the description of the plan should include a rationale for selecting or excluding a specific age range of children. When included, the plan must also describe the expertise of the investigative team in working with children, the appropriateness of the available facilities to accommodate children, and the inclusion of a sufficient number of children to contribute to a meaningful analysis relative to the purpose of the study. If children are excluded then provide appropriate justification. Provide target accrual for this population. See section: Children
Plans for Recruitment/Screening/Retention
See section 8.1.3
Risks/Benefits
Describe any potential risks. This includes physical, psychological, social, legal or other risks. Describe the planned procedures for protecting against or minimizing potential risks and assess their likely effectiveness. Where appropriate, discuss plans for ensuring necessary medical or professional intervention in the event of adverse effects to the subjects. Discuss the potential benefits of the research to the subjects and others. Discuss why the risks to the subjects are reasonable in relation to the anticipated benefits to subjects and others. Discuss the importance of the knowledge gained or to be gained as a result of the proposed research and why the risks are reasonable in relation to the knowledge that reasonably may result.
NOTE: If the study involves the collection, storage or analysis of genetic information, the Genetic Information Nondiscrimination Act (GINA) is invoked. GINA language must be included in the subject consent form. Language can be found in the IRB consent template located on our forms page.
Confidentiality
Provide a statement describing the extent to which confidentiality of materials (data and specimens) identifying the subject will be maintained and that notes the possibility that the Institutional Review Board and regulatory authorities may inspect the materials.
Data Safety and Monitoring
The specific design of a Data and Safety Monitoring Plan (DSMP) for a protocol may vary extensively depending on the potential risks, size, and complexity of the research study. For a minimal risk study, a DSMP could be as simple as a description of the Principal Investigator’s plan for monitoring the data and performance of safety reviews or it could be as complex as the initiation of an external, independent Data Safety and Monitoring Board (DSMB).
The data and safety monitoring plan should provide for a regular review of accrued research data and other relevant information to ensure the validity and integrity of the data and that there is no change to the anticipated benefit-to-risk ratio of study participation. In addition, there should be an ongoing review of study procedures to ensure that the privacy of research subjects and the confidentiality of research data has not been violated.
The following general principles should be considered when addressing an appropriate data and safety monitoring plan:
- Protocols with interventions require some level of monitoring;
- Monitoring should be commensurate with risks;
- Monitoring should be commensurate with the size and complexity of the study;
- Monitoring should be performed on a regular basis;
- Conclusions of monitoring should be reported to the appropriate individuals/groups.
Specific monitoring requirements may be necessary for the following: NIH grant applications for Phase I/II/III clinical trials; Clinical Research Center protocols; University of Vermont Cancer Center protocols; NCI-funded clinical trials; gene therapy trials; or multi-center trials when UVM is the lead institution.
A Data Safety and Monitoring Board (DSMB) is an external, independent committee of scientists, physicians, statisticians, and others that collects and analyzes data during the course of a study to monitor for adverse effects and other trends (such as an indication that one treatment is significantly better than another, particularly when one arm of the study involves a placebo control) that would warrant modification or termination of the study or notification to study participants about new information that might affect their willingness to continue in the study. Typically, protocols that are industry sponsored accrue multiple subjects at multiple sites and are required to have the appropriate resources to capture and report issues of safety and do this by means of a board or equivalent.
Unanticipated Problems to Subjects and Others Reporting
All protocols should specify that, in the absence of more stringent reporting requirements, the guidelines established in the Committees on Human Research “Unanticipated Problems Reporting Policy and Procedures” will be followed. The UVM/UVM Medical Center requirements for reporting adverse events and unanticipated problems to subjects and others should be included in the DSMP. For research protocols utilizing the UVM 3T research magnet, you must have a plan for handling incidental findings. Refer to guidance on incidental findings, see Incidental Findings in Neuroimaging Protocols – Detection and Management policy.
For protocols using the CRC, additional adverse event reporting mechanisms exist. The CRC Office of Research Subject Advocacy is available to assist you in meeting these requirements. Please call the RSA Office (847-0433) or visit the CRC website for sample language on this topic to include in your protocols.
Sources of Materials
Identify sources of research material obtained from individually identifiable human subjects in the form of specimens, records or data. Indicate whether the material or data will be obtained specifically for research purposes or whether use will be made of existing specimens, records or data.
Collaborating Sites
When research involving human subjects will take place at collaborating sites or other performance sites when UVM/UVM Medical Center is the lead site, the principal investigator must provide in this section a list of the collaborating sites and their Federalwide Assurance numbers when applicable. Sites added after initial approval must be submitted as a modification to the IRB and must adhere to the same requirements. Additional agreements may be required.
Consent and HIPAA Authorization
The consent form with incorporated HIPAA authorization language (if protected health information (PHI) is included, (see sections on Consent and HIPAA for further information about PHI)) should accompany the protocol as an appendix or attachment.
References
Include references.
8.1.2 Requirement to include a full Protocol Title in the Electronic Medical Record (EPIC)
UVMMC requires that certain information (full protocol title, CHRMS/CHRBSS protocol number, Principal Investigator and Principal Investigator’s contact information) about research study participation be included in the Research Flag area of each participant’s electronic medical record. This requirement is for participant safety and billing compliance.
It is expected that full protocol titles are included in EPIC, however it is recognized that there may be rare circumstances in which inclusion of the full study title is inappropriate.
If the Principal Investigator feels that including the full protocol title in the EPIC record is not in the best interest of the participant, they may request a waiver of this requirement. The waiver request will either be that the protocol title is withheld completely (EPIC will indicate “Protocol title withheld due to the confidential nature of the research”), or that an alternate title, proposed by the Principal Investigator, is substituted.
Examples of appropriate justification to waive the requirement to include the full protocol title or alter the title:
- There is more than negligible risk of stigmatization or discrimination by health care providers, health insurance plans, employers, or others by placing the protocol title in the medical records of participants.
- There is an expected deterrent effect on research participation by including the protocol title in the medical records of the study population.
- There is misleading information (for example disease condition, drug names) that might lead to conclusions about the person’s condition or related treatment which may increase potential for risk to the participant.
Example of inadequate justification to waive the requirement to include the full protocol title or alter the title:
- Participant privacy by itself is not considered an appropriate justification as electronic medical records are considered private. All personnel with access to the Research Flag in EPIC have completed training in confidentiality and privacy matters and the appropriate access and use of patient medical information. In addition, UVM Medical Center tracks all EPIC access and audit trails are in place to monitor access.
8.1.3 Plans for Recruitment/Screening/Retention
Revised 10.12.2023
The FDA, OHRP and the UVM IRB considers direct advertising for potential research participants to be the start of the informed consent and participant selection process. All methods for participant recruitment, screening and retention need to be reviewed by the IRB. This review ensures respect for persons and their privacy, equitable selection, that the plans and information accurately portray the protocol, and the method is free from coercion. Any changes to the recruitment content must be re-reviewed by the IRB if the platform (newspaper ad, social media account, Front Porch Forum) hosting the advertisement requires changes due to brevity. It may be helpful to reach out to the recruitment platform prior to submitting to the IRB to ensure the ad material can be adequately conveyed to the potential participants without jeopardizing the portrayal of the protocol.
Screening, Recruiting, or Determining Eligibility 46.116(g) and 45 CFR 164.512
The IRB may approve a research proposal (46.111(g)) in which an investigator will obtain information or biospecimens for the purpose of screening, recruiting, or determining eligibility of prospective participants without the informed consent of the prospective participant or the participant’s legally authorized representative, if either of the following conditions are met:
1. The investigator will obtain information through oral or written communication with the prospective participant or legally authorized representative, or
2. The investigator will obtain identifiable private information or identifiable biospecimens by accessing records or stored identifiable biospecimens.
HIPAA under 45 CFR 164.512(i)(1)(ii) Work Preparatory to Research also allows for a researcher to obtain PHI as necessary to establish whether or not it is feasible to proceed with research.
Both of these activities are preparatory to research and must be included as part of your protocol for IRB review and approval. The IRB will review the entire research proposal to ensure that all of the IRB approval criteria at 46.111 is satisfied, including that when appropriate, there are adequate provisions to protect the privacy of participants and to maintain the confidentiality of data at 46.111(a)(7).
This pre-research activity is not a formal waiver of the consent/HIPAA requirement but rather an exception/partial waiver to the requirement until such time as participants are approached to participate in the research project.
Investigators may review existing medical records or biospecimens and they may also approach potential participants to ask them questions to determine eligibility without written consent. However, participants will need to sign consent/HIPAA prior to any procedures as dictated by the protocol for eligibility (e.g. new blood tests, urine tests, imaging.)
Allowable Recruitment Strategies
• Study investigators who are health care providers (clinicians) providing direct care can recruit their own patients
The process needs IRB approval prior to use. Any UVM Medical Center clinician, his/her immediate practice group, or nurses/staff working with those clinicians may approach a patient for research participation.
• Study investigators with no treatment relationship may send a “Dear Doctor” letter asking for referrals of eligible patients who are interested in research participation
The referral letter and the process needs IRB approval prior to use. This recruitment strategy requires that the potentially interested patient contact the researcher as the study investigator has no relationship to the patient. The study investigators are prohibited from having access to patient names, addresses or phone numbers; patients must initiate contact.
• Study investigators with no treatment relationship may provide their colleagues with a “Dear Patient” letter describing the study.
The “Dear Patient” letter and process needs IRB approval prior to use. A PI may send a letter to clinicians requesting that they address and send a “Dear Patient” letter describing the research study to potentially eligible patients. This letter is signed by the treating clinician and provides the patients with contact information for the study investigators. The study investigators are prohibited from having access to patient names, addresses or phone numbers; patients must initiate contact.
• Study investigators with no treatment relationship who are collaborating with the treating clinician may send a collaborative “Dear Patient” letter describing the study.
The “Dear Patient” letter and the process needs IRB approval prior to use. In the event that the research is a collaborative effort between the treating clinician and the study investigators, a joint letter, signed by both, introducing the study investigator as a collaborator along with a description of the study may be acceptable. This letter would inform potential patients that there is an established relationship between their treating clinician and the study investigator.
The preference would be that the interested patient contact the researcher, however in this instance, given that a relationship between their treating clinician and the study investigator has been communicated to the patient, the investigator may contact the patient. A script of the phone contact by the study investigator will be required for review.
• Study investigators with no treatment relationship recruiting patients (inpatient or outpatient)
An investigator may only approach a patient about participation in a research study after permission from the patient to be approached has been documented by the treating clinician, preferably in the medical record.
Study investigators may provide the treating clinicians a study information sheet to be given to the patients.
• Study investigators recruiting potential participants who are unknown to them for behavioral or non-clinical research
Examples include snowball sampling (existing study participants recruit future participants from among their acquaintances), use of social networks, direct approach to unknown people in public situations, and random dialing.
UVM Psychology Participant Pool is an approved mechanism to facilitate undergraduate student participation in research for either required course credit or optional course extra credit. For more information regarding this method of recruitment, contact the Psychology department.
• Advertisements
All recruitment materials including recruitment letters, posters, newspaper ads, radio spots, TV commercials, departmental research websites, or public service announcements are to be forwarded to the IRB for review and approval prior to use. Generally, advertisements used to recruit research participants should be limited to information that a potential participant would need to determine if they are eligible and interested in participating. More specifically, the ads should include information such as:
a. Name and address of the research facility;
b. The condition or disease that will be the focus of the research;
c. A clear statement that the study is research; (required)
d. Summary of criteria for eligibility to participate;
e. Time and commitments that will be required of the participant;
f. Location of the study and the contact for information.
If the tool is a public-facing, departmental research website that lists multiple available research protocols, then you must submit for each individual protocol the content of what you wish to place on the website. We recommend that with each new protocol submission that you determine prior to submission if the protocol will be included in the list of available research protocols and provide the content for IRB approval.
Below are considerations and best practices guidance for all types of advertisements and additional requirements for FDA-regulated research.
The ads should not:
a. Contain explicit or implicit claims of safety and efficacy or equivalency or superiority to other approved treatments.
b. Emphasize the amount of reimbursement that participants will receive. The ads may state that reimbursement may be provided.
For example, posters in which the compensation amounts are too large or are too prominently presented (i.e. bold typeface or large fonts) will not be approved;
c. Promise a favorable outcome or benefit.
d. Promise “free treatment” when the intent is only to say participants will not be charged for taking part in the research.
e. Use terms such as "new treatment," "new medication" or "new drug" without explaining that the test article is investigational.
Recruitment materials should be placed in areas which allow for equitable recruitment of participants. You need to indicate where the material will be placed.
For example, if a researcher advertises in the classified section of the newspaper, the personal column or a “block ad” is considered most appropriate. These should never be placed in the “employment section” of any type of media, (e.g. newspaper or Craig’s list). Particular attention should be paid to emphasize the “volunteer” and “research” aspects associated with participation.
If recruitment is media-based, provide script, if available, and what stations will air it.
If there is a national campaign, provide the press release as soon as it is available and list which stations will air the release.
Recruitment mailings to participants’ should be stamped confidential or personal. We recommend the use of window envelopes to avoid errors in mailing.
Phone recruitment scripts need to be submitted for review and approval. Conversation with parties other than the participant you are trying to contact should not reveal the purpose of the call. Phone mail messages revealing the purpose of the call should be avoided.
It should be carefully explained to a potential participant that voluntary participation in a research project does not constitute employment.
• Direct Mail Recruitment
Direct mail campaigns obtain participants’ names and contact information through large marketing firms who have conducted voluntary surveys of U.S. households. The collected information and consent of the survey volunteer to receive information are placed in a database. This information is then used by direct mail vendors to alert these individuals of new offers or information pertaining to their selected responses.
Details about the mail campaign and the proposed letter and/or materials must be reviewed and approved by the Committee prior to vendor distribution. The letter and/or materials must contain local information such as PI, address, and a telephone number for the participants to contact. There should also be mention of the how the participants’ contact information was obtained for the mail campaign.
• Direct E-Mail Recruitment
Use of a company or university list-serve for recruitment purposes must be approved by the institution that manages/owns that list-serve. The IRB will request proof that permission has been granted by the institution.
• Considerations for Using Social Media to Recruit Research Participants
Terms of Use state the rules of the website on a range of possible issues, including what types of interactions are expected and tolerated on the site, how personal information shared over the site may be used, and who will have access to that information and for what purposes, among other contractual expectations. It is the responsibility of the research team, when designing a protocol, to understand the social media site terms of use. Researchers should be aware of any research or recruitment-related restrictions on the social media sites through which they intend to conduct their recruitment activities. This includes a site’s advertising, privacy and prohibited content policies.
Direct social media (Facebook, Instagram, TikToK etc.) paid ads that do not involve direct communication with potential subjects, can be submitted and reviewed according to existing IRB policy under the Advertisements heading above.
Interactive social media where the study team develops an account specific to the recruitment and retention of study participants is also allowable with prior IRB review and approval. This type of recruitment involves a two-way communication between the participant and the study team through direct messages on social media platforms. Active social media accounts may be best suited for retention of study participants enrolled in long term protocols.
Recruiting via Public and Private Groups
The UVM IRB does allow for recruiting potential participants through both public and private groups set up on social media platforms such as Facebook, Instagram, Reddit etc. Researchers must be aware of any site restrictions on recruiting participants via groups. Researchers must identify if there is a group moderator and request permission to communicate with and recruit group members. It is acceptable to take this step prior to requesting IRB approval. As part of the IRB approval process, researchers must submit to the IRB the text of the recruitment materials that will be shared with group members and their protocols should note any previously obtained approval from the group moderator, plans to obtain approval from the group moderator, or absence of a group moderator. Researchers should consider the process for responding to messages from group members. It is best to direct interested participants to talk questions and information through the private messaging functions or direct individuals to a separately secured email box.
Use of Amazon Mechanical Turk as recruitment venue for surveys and other studies
The use of Amazon Mechanical Turk as a recruitment method for human participant studies continues to grow. Mechanical Turk is advertised as a “marketplace for work,” and individuals who take part in the activities called “HITS” on this site are referred to as “workers.” The compensation for the tasks accomplished is typically very small, usually less than $1. The considerations for using this site for recruitment of participants are the same as with any human participant research. Additionally, the IRB suggests that investigators consider the following:
• Explicitly mention that the study is “research” and not a “job.”
• Address whether or not the compensation is contingent upon certain conditions. Ensure that the complexity of the task and the amount of time expected for completion is reasonable and communicated clearly in the consent process.
Data collected using the Amazon Mechanical Turk data collection tool resides on the Amazon servers and no assurance can be made as to its use for purposes other than the research. The UVM IRB recommends using the below confidentiality statement within the information sheet when collecting data using MTurk.
“All information collected about you during the course of this study will be stored with a unique de-identified code. Work performed on Amazon Mechanical Turk (MTurk) can be linked to your public Amazon profile page, which you control in your sole discretion. Your participation in the research is linked to your profile but not your responses. Thus, you may wish to restrict what information you choose to share in your public profile. More information about MTurk’s Participation Agreement can be found here.”
• Students and Employees
Though the research may be careful to avoid potentially coercive behavior, the very nature of the relationship with the participant can create the appearance of coercion. Even subtle cues of compromise can place participants in a position of involuntary participation in a research project. For this reason, researchers should be aware of the potential for coercion that exists when a research participant is also a student, employee, colleague, or subordinate of the researcher.
Various procedures have been suggested to reduce the possibility of unintended coercion, while still permitting their participation as participants in research. These include:
• Posting IRB approved advertisements/posters throughout the university to recruit participants from a broad base;
• Avoiding any personal solicitations of students by faculty, graduate assistants, or fellow students. In seeking potential participants among employees, the best strategy is to utilize an impartial witness unassociated with the work relationship;
• Providing a number of research projects from which to choose, if participating as a participant is a course requirement;
• Providing alternative and equal methods for meeting course credit (or extra credit) requirements, such as attending a series of research presentations by faculty, writing a brief paper, conducting one’s own research;
• Making it clear in the consent form that refusal to participate will not affect class standing, grades, status on an athletic team, or job standing.
It is IRB practice not to approve recruitment procedures that include employees from the investigator’s own lab or office, especially when the procedures are more than minimal risk. The IRB, however, may reconsider this practice on a case-by-case basis.
Investigator Self-Experimentation
Some researchers may want to participate in their own studies, a practice known as “self-experimentation.” The federal regulations are silent on this point, making no distinction between self-experimentation and participation by others. The IRB requires that such self-experimentation be fully described in a protocol that is submitted for IRB review.
This policy (1) may protect researchers from unwarranted risks and (2) allows a neutral impartial witness to raise concerns, if any, regarding credibility of resulting data.
Note: There is a difference between being a participant in the research and research development/evaluation or testing designed to validate tools for the research project. If the investigator is involved in development only, this would not be considered research and therefore a research study consent form does not apply. However, if any of the procedures involves more than minimal risk, a consent or acknowledgement of understanding should be conducted and documented within the research files.
A main concern for the IRB when reviewing a protocol that involves self-experimentation, is that the ideation of a novel concept may outweigh the investigator’s concern for his/her own welfare.
The IRB may institute additional safeguards for the research project, such as shorter review periods, monthly progress reports, or require that an IRB member obtain informed consent from the investigator.
Screening Options
• Review of patient medical record to determine eligibility
A researcher who is an employee or a member of the covered entity’s workforce could use protected health information to identify prospective research participants under the Preparatory Research provision under HIPAA Privacy Rule at 45 CFR 164.512 (i)(1)(ii). While those individuals may identify potential participants, they should only reach out to those individuals if a treating relationship exists. If no treatment relationship exists, reference the Allowable Recruitment Strategies noted above.
Further IRB review and approval would be necessary to allow removal of information or to further contact potential patients. We would expect this information to be part of the submitted protocol materials.
• Telephone Screening for Eligibility
The practice of telephone screening to establish eligibility is an allowed process. If patients are the population, only those study investigators who are also the treating clinicians or study investigators who have an established relationship to the participant can contact the patient by telephone. The investigator must submit a telephone script for review.
Participant Retention
It is understood that many studies require long-term follow up for disease and survival data. The protocol should account for this follow up from the outset and participants should be made aware of this requirement at the time of consent to participate.
The Committee generally discourages use of participant locator services. Contact by a service rather than the treating physician/researcher may be potentially upsetting to the participant and participants appear to lose their right to privacy when a service, unbeknownst to them, is using different methods to locate them. Requests for use of a locator service are considered on a case-by-case basis and will only be approved if the methods are appropriate and the need for finding a participant is justified.
Any items such as money, small tokens, gift certificates, etc., which are given to the participant to retain their participation in research is considered a form of compensation and needs prior approval by the IRB.
8.1.4 Participant Compensation
Revised 12.12.2023
Compensation may be in the form of money, course points, travel expenses, gift cards, etc. The compensation for participation in the research will be reviewed on a protocol-by-protocol basis. The amount should be commensurate with what is being asked of the participants and cannot be considered coercive. Compensation should not be dependent upon completion of the protocol and there should be a proration schedule. Participation cannot be required for academic course credit or course completion. Researchers should consider how compensation could impact participant’s state or federal benefits (i.e., SSI, SSDI) eligibility in their consideration of compensation type and amount. It is important to note, Principal Investigators are not allowed to use their own personal funds to compensate participants.
Compensation Guidance when Minors are Involved
The level of risk to which a child is exposed is an important consideration for the IRB in determining the appropriateness of payment. The IRB shall ensure that the amount, type, and timing of payment does not unduly pressure or influence the decision making of parents or legal guardians to enroll their child in the research activity.
In pediatric research, inducements are generally tailored to the child participant. Inducements may also be made to the parents or legal guardians of children taking part in research. For example, researchers may provide a payment that represents partial compensation to parents for their time away from work for example when a research study visit requires a full day of the parent's time to accompany their child for a research visit.
Inducements to children shall be age appropriate and respectful and sensitive to children and families. Researchers providing inducements to children shall be encouraged to have several options available for children and families that allow children and families to choose an inducement that is consistent with the family's values. The IRB encourages non-cash payments, e.g., gift cards/certificates, movie/event tickets, toys, books, as forms of payment that are respectful to children and reduce the potential for unduly influencing the child's legal representative's decision regarding participation.
Compensation from UVM Funds
All compensation to research participants is considered IRS 1099 Misc. reportable. Research participants who are eligible for compensation (in addition to cash, this includes gifts, tokens, gift cards and gift certificates) through UVM will be required to provide their name and address each time they are paid. They will also be required to provide their social security number if the amount of the payment is $100 or if the total payments from UVM (all studies) are equal to or greater than $600 in a calendar year. It is important to remember that an individual may participate in a number of UVM studies throughout a calendar year. This is particularly true when the participants live in the greater Burlington area.
If the participant is not a US Citizen or Permanent Resident Alien, they will be required to complete additional paperwork including their immigration status for payment. This information will be strictly confidential and will be used for tax withholding and reporting purposes only and will allow the University to determine US residency for federal income tax purposes. Please contact taxadmin@uvm.edu with questions.
Participant information is collected on the Payment Acknowledgement Form.
If collection of a social security number is required, please us the Payment Acknowledgement Form / Substitute W9. This form only needs to be completed once. Payment Acknowledgement Forms with Social Security Numbers should be transmitted to the Disbursement Center via file transfer. For additional information and detail about processing participant payments and or Payment Acknowledgement Forms, contact the UVM Disbursement Center PurCard team at purcard@uvm.edu.
Companies such as Amazon Mechanical Turk, Prolific or CloudResearch are responsible for collecting the correct tax information and sending out the appropriate IRS 1099 to workers. For research using these tools, a blanket exemption to the collection of Payment Acknowledgement forms is granted. Protocols with an information sheet or consent form using the above survey platforms are not required to add in the UVM procurement language. Please include a usage report with the PurCard journal documentation along with the company invoice.
For other very low dollar compensation, please contact purcard@uvm.edu to seek a waiver on the Consent Template Language. The PurCard team will need to understand the maximum amount of compensation an individual can earn in a year and geographic location of the participants.
Compensation from UVM Medical Center Funds
Research participants who are eligible for reimbursement from the research study through UVM Medical Center must provide their social security number at the first visit regardless of the amount of payment. UVM Medical Center assigns a unique ID upon the first payment visit and uses that ID going forward to reimburse participants.
In both cases all correspondence should be sealed in an envelope and marked confidential.
Compensation from an External Institution’s Funds
When compensation for a UVM research protocol is distributed directly from an external, third party such as a collaborating institution, the researcher must obtain and follow that institution’s policies regarding collection of personal information for tax withholding and reporting. This information should be included in the consent form or information sheet in lieu of the UVM procurement language.
8.2 Chart Review Protocol
Revised 10/02/10
The 2018 Common Rule change in the exemption criteria allows chart reviews to undergo an exempt determination. Researchers now submit through the electronic system under Exemption Category 4iii– Secondary Research Uses of Identifiable Private Information . See section 3.4 Exempt Determination for additional information.
8.3 Qualitative Research Protocol
The “Qualitative Research Protocol Form” should be used instead of the “Human Research Protocol” form when submitting qualitative research or primarily qualitative research and when medical procedures are not included in your research.
Qualitative Research Protocol
The Qualitative Research Protocol form SHOULD be used for proposals where the significant majority of the research methods are qualitative. It SHOULD NOT be used for studies involving human specimens or medical procedures even if the study is primarily qualitative because of the additional regulatory requirements for human subject protection.
Proposals that primarily are composed of qualitative methods, e.g., research “in the field,” phenomenological or ethnographic research proposals may not fit a traditional research design or IRB review model. Still, from the standpoint of federal regulations and professional ethical codes in the social sciences, the same principles and guidelines for protection of the rights and welfare of subjects/participants apply. The IRB has developed a specific “Qualitative Research” protocol form to assist with member review.
You must accurately determine if what you are proposing is qualitative research. Qualitative research is the gathering of data primarily through the methods of participant observation, observation, face-to-face interviews, or open-ended surveys or questionnaires, often for the purpose of addressing questions pertaining to social and/or cultural issues. There are several kinds of qualitative research, such as ethnography, historical, field research, phenomenological, grounded theory, and case studies. Such research may describe a social situation as a whole, or it may focus on specific problems or situations within a larger social context.
Quantitative research generates numerical data or information that can be converted into numbers.
Medical research means direct medical intervention or interaction, clinical trials for new drugs/devices, FDA regulated activities, invasive or non-invasive medical procedures for research purposes or the collection or use of private health information for research purposes in the biomedical arena.
Sometimes research protocols combine both qualitative and quantitative research methods, referred to as mixed-method research. If you are doing mixed methods research with equal parts qualitative and quantitative methodology, you will need to choose the submission form that best describes your research to someone outside of the study team (i.e. for IRB staff and Committee Members to review).
The IRB appreciates that qualitative research has the following special characteristics (Arwood, T., and McGough, H., 2007 PRIM&R SBER Conference):
- It is experiential
- It is interactive
- It is not easily bounded by time and place
- It is often exploratory
- It morphs easily and often (new questions emerge during research)
- The boundaries between normal activities and communication and data collection are blurred
In order for the IRB to understand these special characteristics and the nature and scope of a particular qualitative research project, the following issues should be addressed in the Qualitative Research Protocol, if applicable.
Participant Population
- The kinds of people who will be involved in the research should be described. If there are different groups or categories of people, the groups and the approximate number of participants in each group anticipated to be enrolled must be described. If potentially vulnerable populations are included, any additional protections should be explained. If an exact number of people to be enrolled are unknown, a range should be provided. A modification should be submitted to the IRB when/if actual numbers exceed those estimates.
Description of Procedures
- The length of time to be spent at the field site(s) should be described. If unsure, an approximate length of time should be provided (e.g., one year, two summer months, etc.). Also the approximate length of time of the interaction with subjects (i.e. 2 hour interviews, day-long observation) and the number of anticipated interactions (i.e. 3 interviews over a 4 month period) should be provided. A modification should be submitted to the IRB when/if actual dates exceed those estimates.
- The research techniques that will be used to conduct the research (such as participant observation, interviews, focus groups, use of public, private governmental or other records, administration of test, etc.) should be described. The topics or research domains to be covered as well as what will be observed (such as individual behaviors, community rituals, societal norms, etc.) should be described. This will help the IRB get a sense of what will be learned from and about the participants in the research.
- Explain how the maximum number of participants is determined or what criteria will be used to determine when data collection is completed.
Research Site(s) or Location(s)
- Explain where the research will be conducted and explain why this particular research setting was chosen.
- Has the researcher conducted research at this site or with the population previously? If so, briefly describe the topics and duration of your previous research..
- Is local governmental or community permission to conduct research required at any of the sites? If so, explain how you will obtain this permission. If there is formal documentation of this permission, attach it to the application form or indicate when it will be received and forward to the IRB.
- Will you work with local collaborators (interviewers, interpreters, translators, guides, etc.)? If so, please explain who these collaborators are and how they will be involved in the research. Will they need to obtain local ethics committee approval for their role in the study?
- Many countries have the expectation that foreign scholars will collaborate with local scholars and institutions. Explain whether this applies to your research and if local IRB or other type of ethical review board approval will be obtained.
Risks and Inconveniences
Risk of harm in qualitative research is usually limited to what may result from invasion of privacy, stigmatization, or breach of confidentiality. Harm may happen to individuals and to the groups or communities to which they belong.
- Identify the risks of harm that may result from this research.
- Describe the steps you will take to minimize the risks of harm. If harm occurs, what plans do you have to manage it?
- If there are different risks of harm for different groups of participants, please identify the risks for each group. Sometimes this cannot be known in advance of entering the field. If unanticipated problems occurrence research has begun, the incidents must be reported to the IRB. When appropriate, the study can be modified to address any issues that arise.
Benefits
- Is it possible that individuals who take part in your research can reliably expect a direct benefit from taking part? If yes, describe.
- Describe the anticipated benefits of this research for the community you will study, for your profession, or for society in general.
Confidentiality
- Describe how you will find out how people in this setting feel about the fact that you will write articles about them. Will you consult with the people from whom you collected data before you publish?
- Are any portions of the research material you may collect not publicly available and expected by community standards to be private? If yes, describe the materials that are private and explain (1) how you will store the private information or materials while you are in the field so that the confidentiality of the data is protected; (2) explain how you will store the private information or materials after you leave the field so that confidentiality is protected; (3) explain whether you will retain information that could lead to identification of the research site and explain any negative consequences this could have; (4) explain if you will record any direct participant identifiers (such as names or contact information) that could be linked to the private research material.
- If you will record identifiers (# 4 above) explain why and describe how you will protect against disclosure of this information or explain why this is not necessary. If you will retain the identifiers linked to the data, explain (1) how long the identifiers will be kept, (2) how confidentiality will be maintained during this period, (3) who will have access to data (such as sponsors, advisors, government agencies, etc.). In each case, explain whether they will have access to study data with identifiers or only to coded data with no access to the identifying study code. If identifiers will be maintained indefinitely, explain why. For example, do you intend to re-contact participants or communicate with them over a long period of time, or is the data identifiable by its nature (recordings, genealogies, etc.). Explain how you will protect the data from a breach of confidentiality or why this is not necessary.
- If you will retain data that may place participants at risk for criminal or civil liability or be damaging to their financial standing, employability or reputation, please explain. It may be advisable to obtain a federal Certificate of Confidentiality.
- The IRB acknowledges that sometimes it is not possible or desirable to maintain anonymity. For example, when a researcher works with a small group of people only found in a particular region with whom others have worked. In order to advance ethnographic knowledge about the group, their identity must be made known.
Sometimes individuals or whole communities do not want to remain anonymous. If this is the case, explain how you learned of this and describe why. If there are differences in the community about this, describe how this will be handled.
Consent Procedures/Process
- Explain how you will introduce yourself as a researcher to potential participants. If you already know them, please explain the circumstances.
- How will you inform people about your research and obtain their consent to participate? If you plan to use an oral consent process and to work informed consent procedures into your introduction to a group, or the beginning of an interview, please provide a general script or a list of points you will cover.
- Describe how people in this setting let you know if they don’t want to talk with you.
- Identify who is responsible for giving consent in the research setting (for instance, if a tribal council or community leader provides consent for the entire group). Sometimes the consent process can be multi-layered in community settings. Be sure to describe what the full process is in the setting in which the research will take place.
- Describe how you will handle situations in which group consent is provided, but individuals to not want to participate and vice versa.
8.4 Biological Specimens/Data Repository Protocols
There is a category of expedited protocols that include the collection of samples or data for future research. What follows is policy and guidance for repository protocols. We have developed a submission form “Biological Specimens/Data Repository Protocol” to address management of repository activities.
NOTE: If you intend to share research data sets with other colleagues, you must obtain local IRB approval as well as the colleague’s IRB approval. The IRB typically requests that all individual identifiers be stripped prior to release. Justification for maintaining identifiers, even if coded, will be required. Sharing data outside of our institutions may require that a data use agreement be obtained. UVM investigators should contact Sponsored Projects Administration at 656-3360 to speak with the Executive Director. UVMMC investigators should contact the Office for Clinical Trials Research at 847-8990.
Biological Specimens/Data Repository Policy
The use and control of human tissue and medical charts for research is governed and restricted by federal and state laws and local regulations to ensure human protection measures are adequate. In the past tissue registries, tissue banks, pathology archives, research waste materials, hospital and clinic charts, and other databases have often been accessible to medical researchers. Often this “tissue” material was acquired from human subjects (living persons and fetuses) for non-research purposes such as diagnosis, medical therapy, public health control, quality assurance and transfusion/transplantation therapy. Researchers were often permitted access to these materials without adequate human protection mechanisms in place. More recently, human protection standards on use of tissue material have become more stringent and less trivial based on newly identified issues such as medical/legal privacy acts, HIV status, genetic confidentiality issues, religious and ethical beliefs, fetal restrictions, and other issues. The days of free access to personal data and tissues by researchers without subject consent have passed.
Operation of a specimen/data repository is now subject to oversight by the committee. The committee will review and approve a protocol specifying the conditions under which sample collection occurs and then all subsequent requests to access the specimens or data for research purposes. This is done to ensure adequate provisions to protect the privacy of subjects and maintain the confidentiality of data.
Definitions
Human tissue and data repositories collect, store, and distribute human tissue materials and data for research purposes. Repository activities involve three components: (i) collection of the tissue samples or data; (ii) storage of the tissues samples or data; and (iii) future use of stored tissue or data.
Tissues include any cell tissue, fluid, or excreta from which measures of normal or pathologic human physiologic function can be obtained. The term, “tissue” includes, but is not limited to pathological specimens, diagnostic specimens, hair and nail clippings, deciduous and permanent teeth, dental plaque and calculus, sweat, uncannulated saliva, placenta removed at delivery, amniotic fluid, cerebrospinal fluid, genetic material, urine, blood and other bodily fluids.
Data includes any private medical or non-medical information obtained from the subject, informant or the medical chart.
Coded means that identifying information (HIPAA Identifiers List) that would enable the investigator to readily ascertain the identity of the individual to whom the private information or specimens pertain, has been replaced with a number, letter, symbol, or combination thereof (i.e., the code); and a key to decipher the code exists, enabling linkage of the identifying information to the private information or specimens.
Protocol Requirements
The IRB has developed a “repository” protocol form and process that covers all the following requirements.
1. person responsible;
2. description of all tissue/data that will accumulate in the bank;
3. physical location and security measures of the bank;
4. a separate “repository consent form” or request for a waiver of consent/authorization;
5. pledge that the responsible party will not use or release tissue or data unless an IRB application is submitted for every proposed analysis of that data;
6. internal monitoring in place to cross check samples, data, consents and withdrawals;
7. safeguards against identification; use of a third party system;
8. process to evaluate tissue utilization;
9. length of time specimens or data are kept; how destroyed; and
10. what information will be shared with subject(s).
As is usual, the protocol should discuss these issues in enough detail for the IRB members to evaluate the plans; a somewhat briefer description should be included in the consent form.
OHRP strongly recommends that a Certificate of Confidentiality be obtained to protect confidentiality of human cell repository specimens and data.
Consent Form Requirements
The IRB has developed a standard specimen collection consent form which includes the following elements:
1. operation and security of the repository;
2. specific types of research to be conducted (as specific as possible with the basic idea to determine if there is any type of research to which the subject might object);
3. conditions under which data and specimens will be released to recipient-investigators;
(other site must have IRB approval);
4. procedures for protecting the privacy of subjects and maintaining the confidentiality of data;
5. sensitivity of the generated information (E.g. information that could affect a subjects employment, insurance coverage etc., (if DNA typing is involved include information on consequences (paternity));
6. non-exculpatory language through which subjects are made to waive or appear to waive any legal rights;
7. freedom to withdraw: how will confidentiality risk be terminated;
8. whether or not it is anticipated that the subject may be approached for follow-up information or follow-up samples in the future (a process to which the subject should have the opportunity to give or deny consent);
9. voluntariness of participation;
10. the degree to which ongoing access to medical records is being sought for correlative information, and the duration of such access; and
11. the kind of information that will be provided to subject(s).
Reporting Individual Results to Subjects
For general repository activities, it is probably best to plan not to provide results of future studies to the subjects. Much of the future research will be conducted without identifiers, and it is unlikely that most research will yield results that, if known, would affect the patients' health care or family planning. In fact, there are many cases in which it would be unethical to provide results to patients, such as when the research is in the early stages and the clinical significance has not been established. On the other hand, if there is a good chance that research will yield results that could affect the subjects' medical care, it may be appropriate to tell subjects that if such identifiable results are obtained, the subjects will be contacted and asked if they wish to be informed of the results. The decision about whether to provide any results to subjects will depend on the ethical implications of each protocol.
Note: Results obtained from a laboratory that does not meet the standards of CLIA (Clinical Laboratory Improvement Act) may not be used to direct patient care. In these cases, the subject could be notified that an issue has been identified, and that the subject or in some cases the investigator can notify the subject’s physician so that their physician may follow-up with standardized testing when appropriate.
Receiving or Purchasing Tissue/Data from Other Researchers
For protocols in which there are plans to collect specimens or data from outside institutions, the committee will review a “collection” protocol (can use same “repository” protocol form), informed consent /authorization document (if applicable) for distribution to these outside specimen/data collectors and their local IRBs.
A written agreement is required for collector-investigators, which requires written informed consent of the donor-subjects utilizing an informed consent document or a waiver approved by each of the local IRBs. It should also contain an acknowledgment that collector-investigators are prohibited from providing recipient-investigators with access to the identities of donor-subjects or to information through which the identities of donor-subjects may readily be ascertained.
Giving Tissue/Data to Other Researchers
IRB review required: A UVM/UVM Medical Center researcher who is responsible for a tissue or data bank cannot release tissues or data to other researchers (local or not local) until he or she has determined that those researchers have received appropriate approval or determination of exemption from a duly constituted Institutional Review Board (IRB).
Researchers at other institutions or companies are not subject to review by UVM/UVM Medical Center IRB, and the IRB has no jurisdiction over how non-UVM/UVM Medical Center researchers will protect subjects' privacy and interests in the future research. Researchers who are funded by private foundations or industries may be conducting research which is not necessarily subject to Federal regulations protecting human subjects. Therefore, to apply the same level of protection for all subjects involved in UVM/UVM Medical Center research, the IRB generally will not permit providing subject identifiers along with tissues or data to non-UVM/UVM Medical Center researchers. If necessary, the samples/data may be coded, but the key must be maintained at UVM/UVM Medical Center. For those University of Vermont or UVM Medical Center protocols in which there are plans to release tissue or data to an outside institution, a written usage agreement for recipient-investigators is required.
Other researchers at UVM/UVM Medical Center wishing to use samples or data previously collected under an approved protocol should contact the IRB office for assistance with determining what paperwork is required for review.
Specimen Collection as Part of a Larger Protocol
Tissues are routinely used for a variety of tests within treatment and epidemiological protocols, and most researchers are accustomed to describing the sampling procedures and risks in their protocols and consent forms. However, if some of the samples will be saved for unspecified future research, additional discussion is needed in the protocol and consent form. All of the concerns that apply to independent tissue collection protocols apply here as well.
Making Tissue Collection Optional: In treatment studies with a potential benefit for subjects, the IRB recommends that collection of tissues for repositories (as opposed to collection for treatment-related analyses) be made optional. That is, in general, patients should not be denied a promising experimental treatment because they are unwilling to allow their tissue to be banked for unspecified future purposes. The IRB is aware that some national cooperative study groups make banking of samples a condition of study participation. Approval for such repositories will be considered very carefully and denied only when there are over-riding ethical concerns.
Consent within a larger protocol: A consent form should emphasize the major procedures and risks of the research study. If the study primarily involves banking of specimens in respositories for future research use, then the major part of the procedures and risks sections should discuss this long term storage. On the other hand, when banking is done as a small and preferably optional part of a treatment study (a cancer treatment regimen, for example) the concern about banking is relatively small compared to deciding how to treat a life-threatening condition.
One acceptable method of discussing banking within a consent form for a treatment study is to include a separate paragraph between the Procedures and Risks section, briefly discussing the banking and its implications. If banking in a repository is optional, two appropriately labeled lines for initials should be included with the signature section, so that subjects can indicate whether or not they are willing to have specimens banked.
The IRB recommends the following language:
“Biologic Studies: Portions of your tissues which are removed as part of care and are not required for routine diagnosis or treatment may be used now or in the future for research purposes. These tissue samples may be used to learn more about how cancer or other diseases develop and/or may result in new products, tests or discoveries. In some instances, these may have potential commercial value. These tissues to be kept for research purposes will be obtained only at the same time as your regular procedures are performed; you will not have to undergo any special procedures for this purpose. There will be no additional charge, and you will not receive any payment or financial benefit from any products, tests or discoveries. You may also be asked in the future if you are willing to be in additional research studies. You will not be told the results of any future research. Participation in this extra research is voluntary, and if you choose not to allow the extra research it will in no way affect your care on the main study. You may at any time contact the researchers to request that your samples be withdrawn from research use, and any identifiable samples still in their possession will be destroyed. Please indicate whether you are willing to allow this extra research by initialing one of the lines at the end of the form.”
Just before the signature lines in the treatment consent form:
“______ I do not want my tissue and blood samples used for any research or tests other than those needed for the main research study.
______ The researchers may keep my extra tissue and blood samples for future research.
______ I am willing to be contacted in the future about any additional research studies.”
8.4.1 Research Tissue Acquisition Policy
UVM’s IRB has adopted the UVMMC’s 6-1-2014 Tissue Acquisition Policy as stated below.
Specimens obtained as part of research protocols:
“Patients sometimes enroll in research studies and agree to have tissue obtained for study purposes. It is essential however, that sufficient tissue be obtained for complete examination in the Department of Pathology to ensure an accurate diagnosis for the patient even when samples are required for research purposes. Therefore, research protocols requiring tissue that would otherwise be sent to pathology for examination must (i) include procedures for ensuring the adequacy of diagnostic tissue for pathologic examination and (ii) receive approval from the surgical pathology quality assurance subcommittee on research specimens prior to activation. Such protocols must acknowledge that if feasible, tissue diverted from pathologic examination for investigative purposes will be provided to the Department of pathology for diagnostic or other patient management purposes as clinically indicated. Pertinent information for patients regarding the use of their tissue for research purposes (e.g. risks and benefits) must be included in the informed consent for such research studies.”
8.5 Blood Collection Protocols 46.110
Revised 10.17.2023
This is a category of expedited protocols that includes only the collection of blood. Researchers conducting bench science often times require human cells for their laboratory research, and they and their staff, will donate blood for these scientific purposes. While the activity of drawing blood is not a “research protocol”, the collection of the cells is for research, and therefore falls under the IRB purview. What follows is guidance for blood collection protocols.
Guidance Blood Collection Protocols
OHRP has ruled that for all Federally sponsored research, informed consent must be obtained before identifiable specimens may be collected for research purposes. This applies to any research that is done at an institution that receives Federal funding. The following guidance and the Blood Collection Protocol form have been developed to comply with these requirements and to help standardize procedures to establish safe practices in the collection of human blood and human blood products for research purposes. Submission of the Blood Collection Protocol for review and approval will assist the IRB in ensuring that donors who are participating are protected.
Blood Collection in Non-Clinical Settings
Blood draws in spaces outside of clinical care areas should be conducted in a room that is separated by a door from bench space, biological safety cabinets or other laboratory equipment that is used to handle or store biological infectious agents. Space utilized for blood draws should be separated from active manipulation of infectious biological agents and active work with hazardous chemical agents prior to the blood draw (for area disinfection purposes), at the time of the blood draw, and until disinfection procedures have been completed after the blood draw.
Blood draw areas must follow all BSL-2 work practices including:
1. Furniture – Blood draw chair or table should be made of materials that can easily be disinfected (example vinyl or plastic furniture)
2. Sharps containers – An approved Sharps disposal container should be available in the blood draw area at the point of use. All glass items and needles must be disposed of in an approved Sharps container.
3. Disinfectant – Bleach solution or an EPA registered disinfectant should be available in the draw area in the event of a spill.
4. Spill or Emergency Procedure - A procedure to handle spill cleanup or emergency response information should be posted at the point of use.
5. Biohazardous Waste Disposal - Biohazardous waste bags and boxes must be used to dispose of all plastic ware and personnel protective equipment.
6. Biohazardous Signage and Labeling – All clinical laboratory spaces must be labeled with a biohazardous door sign designating the space as BLS-2. All equipment used to store and handle human blood and blood products must be labeled with a biohazardous sticker.
7. Personnel Protective Equipment – Personnel conducting blood draws are required to wear the appropriate personnel protective equipment (PPE). This includes liquid barrier gloves (latex or nitrile), face protection (full face shield or surgical mask and safety glasses) and lab coat or lab gown that can be laundered or disposed in event of a blood splash or spill.
Personnel Conducting Blood Draws
The principal investigator is responsible for verifying that personnel performing blood draws have sufficient training and experience in conducting human blood sampling. Qualifications may include prior experience as a trained phlebotomist, nurse or emergency medical technician. All research personnel conducting human blood draws or work with human blood and blood products must complete blood borne pathogen training on an annual basis. Information on training is available on the University Environmental Safety website or is part of the UVM Medical Center mandatories.
Approved Standard Practices for Obtaining Blood
1. Healthy individuals will be asked to participate in this minimal risk procedure. Education and review of the consent will be performed.
2. After the consent is signed, the volunteer will be brought to ______________________
3. Phlebotomy of a peripheral arm vein will be performed by ________________________using sterile procedures and seated position. A sterile bandage will cover the phlebotomy site after the procedure and the arm will be elevated to ensure that bleeding has stopped.
4. The volunteer will be observed for any lightheadedness, bruising or bleeding during and after the procedure.
5. If the volunteer is lightheaded, he/she will be reclined and monitored until symptoms resolve.
6. If the volunteer is asymptomatic after the phlebotomy procedure, he/she will be released.
7. Any volunteer with any side effects during or after phlebotomy will not be used again to obtain the blood products. In addition, any volunteer that requires more than three attempts to access a vein will also not be used as a volunteer.
8.5.1 Blood Drawing Limits
Revised 1/14/22
As a general rule, investigators must not draw more blood from any research participant than is
needed to answer the research question, and should design the research to minimize that
volume. Investigators are strongly encouraged to obtain research blood at the same time as any
clinical labs are being collected, if possible. Good clinical decision making should always be used to ensure the safety of the research participant.
A. Blood Drawing Limits for Protocols Reviewed Using the Expedited Procedure
Collection of blood samples by finger stick, heel stick, ear stick, or venipuncture as follows:
- from healthy, non-pregnant adults who weigh at least 110 pounds. For these subjects, the
amounts drawn may not exceed 550 ml in an 8-week period and collection may not occur
more frequently than 2 times per week.
- children, considering the age, weight, and health of the subjects, the collection procedure,
the amount of blood to be collected, and the frequency with which it will be collected. For these subjects, the amount drawn may not exceed the lesser of 50 ml or 3 ml per kg in an 8 week period and collection may not occur more frequently than 2 times per week.
- Depending on the health status of the research subject, the IRB may require the investigator to justify the need for volume of blood removed in relation to the expected benefit to the participant and safeguards to protect from undue risks.
B. Blood Drawing Limits for Protocols Reviewed by a Convened Board
The convened IRB may approve a volume of blood drawn for research purposes that exceeds
the limits referred to above. As a general rule, blood drawn for research purposes must not
exceed the following volumes:
- For an adult, the amount of blood that may be drawn for research purposes shall not exceed 5
mL/kg in any one 24- hour period, and 7 mL/kg in any eight- week period. Any exception to
these limits must be specifically justified in the research protocol and approved by a convened
IRB.
For a child, no more than 3 ml/kg in 24 hours with perhaps up to 5 ml/kg over 8 weeks. This assumes an otherwise healthy child. Research samples should be drawn at the same time as clinical ones if possible. Any exception to these limits must be specifically justified in the research protocol and approved by a convened IRB.
- Some pharmacokinetics studies may exceed the blood volumes listed above. These requests will be reviewed by the committee on a case by case basis.
8.6 Research Involving Coded Private Information or Biological Specimens
Basic Policy
Research using private information or biological specimens that is categorized as “human subjects research” can only be conducted after receiving the appropriate Committee review under the normal established guidelines.
There is now federal guidance that clarifies the distinction between (a) research involving coded private information or specimens that does not involve human subjects; and (b) human subject research that is exempt from the requirements of the Department of Health and Human Service (DHHS) regulations. The guidance also provides new definitions of key terms to assist in making these determinations because it fits into specific categories. When conducting research using data or specimens, the human subject determination noted above, the type of application, the informed consent requirements, and the level of review by the IRB depends primarily on one factor: whether the data or specimens are identifiable to the principal investigator or key personnel.
Determinations of whether research involving coded private information or biological specimens is considered to be “human subjects research” must be made by the IRB, not the investigator.
Does Your Proposed Research Involve Human Subjects?
Regulatory requirements (Federal and state) to protect human subjects apply to a much broader range of research than many investigators realize, and researchers using data and/or specimens are often unsure about how regulations apply to their research. Regulatory obligations to protect human subjects would apply, for example, to research that uses –
1. Bodily materials, such as cells, blood or urine, specimens, organs, hair or nail clippings, from living individuals who are individually identifiable to the investigator(s), even if these materials were collected by others;
2. Residual diagnostic specimens from living individuals that are individually identifiable to the investigator(s), including specimens obtained for routine patient care that would have been discarded if not used for research;
3. Private information, such as medical information, about living individuals that is individually identifiable to the investigator(s), even if the information was not specifically collected for the study in question. This includes research on genetic information that can be readily associated by the investigator(s) with identifiable living individuals.
The definition of “human subject” includes, but is not limited to, human organs, specimens, and body fluids from living individuals, as well as private graphic, written, or recorded information about living individuals, if (1) there is interaction or intervention with a living individual to obtain the data or specimens for research purposes, or (2) the identity of the subjects can be readily ascertained by the investigator or other members of the research team.
Research that involves only coded private information/data or coded human biological specimens may not constitute human subjects research under the HHS human subjects regulations (45 CFR Part 46) if:
1. The private information and/or specimens were not collected specifically for the currently proposed research project through an interaction/intervention with living individuals, AND
2. the investigator(s) (including collaborators) on the proposed research cannot readily ascertain the identity of the individual(s) to whom the coded private information or specimens pertain (e.g., the researcher’s access to subject identities is prohibited by written repository procedures and policies and/or through an agreement signed between the recipient researcher and the repository providing the data or specimens).
Individuals who provide coded information or specimens for proposed research and who also collaborate on the research involving such information or specimens are considered to be involved in the conduct of human subjects research.
The same data/specimens may be identifiable or not for different researchers. For example, if Researcher A stores or receives data/specimens with a code number and has a key linking the numbers to subjects' identities, the data/specimens are identifiable for Researcher A. On the other hand, if Researcher A passes the data/specimens to Researcher B and there is a written agreement that Researcher A will in no circumstances give identifying information to Researcher B, then for Researcher B's research the data/specimens are not identifiable and not considered research involving human subjects.
Who Determines Whether Human Subjects are Involved in Research
The decisions about when research involving data and/or specimens from subjects is considered human subjects research are complex. The project must be submitted to the IRB for this determination utilizing the form titled “Research Not Involving Human Subjects Review and Determination .”
Retrospective versus Prospective Collection of Data or Specimens
The criteria the IRB must use to determine whether research involves human subjects under this policy are based on the following types of data/specimen collection methods and how the data/specimens are identified.
- Retrospective data or specimens are data or specimens that are already existing or "on the shelf" when the research is proposed. Specimens or data are considered existing only if they were gathered before the research is proposed. Example: Data previously collected for medical care or specimens that are left over from surgery or previous research studies.
- Existing, Not Identifiable (i.e., not coded) data or specimens: This category includes data or specimens obtained without identifiers from a data or specimen repository at UVM/UVM Medical Center or elsewhere. Research utilizing such existing coded data or specimens would not be considered “research with human subjects. ” The investigator would need to complete and submit the “Research Not Involving Human Subjects Review and Determination” form.
- Existing, Identifiable data or specimens: This category includes data or specimens that can be individually identified with the patient/subject and therefore would be considered “human subject research. ” This type of research may meet the federal criteria for exemption from IRB review. See guidance on exempt review in the Research Manual.
- Prospectively gathered data or specimens: Conversely, data or specimens that will be taken from patients or subjects after proposal of the research are considered to be prospectively gathered.
- Prospectively Gathered, Not identifiable (i.e., not coded) data or specimens: If you are prospectively obtaining data or specimens which are left over from another purpose (clinical, diagnostic procedures or another research study), and they are not individually identifiable (coded) to the investigator, it may be considered not “research with human subjects.”
- Prospectively Gathered, identifiable data or specimens: If the data or specimens are to be gathered specifically for the research project, it is considered “human subject research” and would not qualify for exemption, however might qualify for expedited review. See Research Manual for guidance regarding expedited review.
8.7 Sustainable Agriculture Research and Education (SARE) Grant Projects
Revised: 12/9/20
Background
The Sustainable Agriculture Research and Education (SARE) program is a competitive grants program that funds research and education projects in every state and island protectorate. Funded by the United States Department of Agriculture's National Institute of Food and Agriculture, the program has four independently-run regions (North- Central, Northeast, South and West) hosted by land grant institutions and guided by regional Administrative Councils comprised of agricultural stakeholders. SARE Outreach provides communication and technical support at the national level.
The Northeast region is hosted by the University of Vermont (UVM). Northeast SARE offers six different types of grants, most of which are open to commercial farmers, educators, researchers, nonprofit organizations, public agencies and businesses. The prime award from USDA NIFA is awarded to UVM with the Director of Northeast SARE as the Principal Investigator. Grants under the prime award are awarded as subawards to institutions and as service agreements to individuals/private businesses, as is the case for farmers.
UVM has an obligation to assure that Northeast SARE’s grantees are aware of the need to comply with IACUC and IRB if their projects involve animals or human subjects. In most cases, grantees work with their home institutions or organizations to make that determination and develop plans as needed. However, small organizations and farmer grantees do not have this capacity.
Scope of Process
This process addresses awards made to individuals and organizations in Northeast SARE’s Farmer Grant, Partnership Grant, Research and Education, Research for Novel Approaches, and Professional Development programs. Whenever possible IACUC or IRB determinations will be made by an official review committee arranged by the awardee.
Projects Involving Animals
The University’s IACUC committee is not able to formally review projects from individuals who are not employees of UVM. However, if SARE receives a grant proposal that includes animals, the SARE program may request a UVM veterinary review of potential impacts on animal welfare. In that case SARE program staff will email the proposal and completed livestock care questionnaire to Dr. Ida Washington, University Veterinarian and Director, Office of Animal Care Management at University of Vermont, at Ida.Washington@uvm.edu. Dr. Washington will provide an independent review and a determination of whether or not the project meets appropriate animal welfare standards. This determination could include: that the project meets animal welfare standards; a request for clarification on aspects of a project; a request for specific changes to be made in project approach; or other determinations as appropriate.
SARE staff will work with the awardee to ensure Dr. Washington’s recommended changes are made prior to release of funds.
For awardees required to provide IACUC review outside of UVM, SARE staff will require that the IACUC review results be provided before research funds are released.
Projects Involving Human Subjects
All SARE grantees, whether UVM or non-UVM employees, with projects that include human subjects research, must either be exempted from IRB review or obtain IRB approval of their research project. Exempt human subject studies present no more than minimal risk to subjects and fit into one or more categories as outlined under 45 CFR 46.104(d). Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. Studies that qualify for exempt status do not have the same federal requirements for research involving human subjects as non-exempt studies.
UVM SARE Grantees
UVM PIs with approved SARE projects that will conduct research that includes human subjects are required to submit their projects to the UVM IRB for either a formal exemption determination or research protocol approval.
Non-UVM SARE Grantees
Non-UVM PIs with approved SARE projects that will conduct research that includes human subjects will have their projects reviewed by SARE staff, who will make an informal determination as to whether or not the project appears to be exempt from IRB review because the human subjects research poses minimal risk, as defined above. To make this determination, SARE staff will reference the materials found on UVM’s IRB website https://www.uvm.edu/rpo/determine-if-project-requires-irb-review, and on the U.S. Department of Health & Human Services website at https://www.hhs.gov/ohrp/regulations-and-policy/decision-charts/index.html#c1.
UVM's IRB is not able to formally review projects from individuals who are not employees of UVM, but SARE staff may contact UVM’s IRB staff for advice on exemption determinations. If IRB staff offers guidance to SARE staff as to how the project can be changed to meet the exemption criteria, SARE staff will require that the grantee modify the proposal accordingly before funding the project, or require that the grantee obtain an IRB review. Which of the two options is most appropriate will be determined on a case-by-case basis to determine the best course of action that supports grantees in implementing their projects safely and in accordance with human subjects protections.
Project PIs notified by SARE staff that they must have an IRB review can obtain that review from their host institution, a project collaborator’s institution, or an external IRB.
Release of funds
For both UVM and non-UVM projects required to have an IRB review, SARE staff will require that documentation of the IRB review results be provided before research funds are released.
8.8 Department of Defense (DoD) Supported Research Projects
New 10/02/10
All awards issued by the Department of Defense are subject to the regulatory policies and procedures of the US Army Medical Research and Material Command (USAMRMC) under DoD Instruction 3216.02, effective April 15, 2020. Researchers may not begin research activities involving human subjects, biospecimens, and/or human data or expend funding on such effort until applicable regulatory documents are reviewed and approved by the US Army Medical Research and Material Command (USAMRMC) to ensure regulations are met.
The USAMRMC Human Research Protections Office (HRPO) is the entity within the DoD that reviews human subject research. Questions regarding applicable human subject protection regulations, policies, guidance should be directed to hrpo@amedd.army.mil.
Scope
These requirements apply if any of the following conditions are met.
1. The research is funded by a component of the Department of Defense (Navy, Army, Air Force, National Geospatial Intelligence Agency, National Security Agency, Defense Intelligence Agency, Defense Threat Reduction Agency, Defense Advanced Research Projects Agency, and United States Joint Forces Command);
2. The research involves cooperation, collaboration, or other type of agreement with a component of the Department of Defense;
3. The research uses property, facilities, or assets of a component of the Department of Defense; or
4. The subject population will intentionally include personnel (military or civilian) from a component of the Department of Defense.
Requirements of UVM
To be eligible to receive DoD awards for human subject research, UVM must have a current OHRP Federalwide Assurance.
Who Is the Reviewing IRB
- If the project is a direct award to UVM from the Department of Defense, the UVM IRB is required to review and approve then a secondary review and approval is carried out by HRPO.
- If UVM researchers are collaborating directly with Department of Defense personnel on a project, the DoD personnel are responsible to obtain the DoD’s IRB approval. UVM has the option to rely on the DoD’s IRB through a reliance agreement. In this circumstance, UVM would not need to review the study. Processes to allow reliance on another IRB are in Section 13.3 of the manual.
- If UVM is a subawardee, UVM may rely on the prime awardee IRB review and approval under a reliance agreement. Processes to allow reliance on another IRB are in Section 13.3 of the manual.
Requirements of the Principal Investigator
Prior to release of DoD funds to conduct research, UVM researchers must:
- Submit the following to the UVM IRB for review (some are DoD requirements):
- Protocol
- Consent with UVM and specific Department of Defense language included in the appropriate sections as listed below:
- A statement that the US Army Medical Research and Material Command is providing funding for the study.
- A statement that representatives from the DoD are authorized to review research records.
- In the event that a HIPAA Authorization is required, DoD must be listed as one of the parties to whom private health information may be disclosed.
c. Documentation of scientific merit through independent scientific review of the protocol, see below Scientific Review;
d. Other items as applicable.
2. Verification of ethics training completion for all staff listed on protocol. This includes the general Human Subjects Training and Good Clinical Practices. The DoD component will compare this to their requirement.
3. If applicable, waiver of informed consent approval from the Secretary, see below Intent to Benefit.
Once you have UVM IRB approval you must submit all of the approved materials to the HRPO following their directions. It is strongly recommended that investigators read the “Information for Investigators”, dated June 2020 located on the HRPO page as time to approval depends greatly on adherence to the requirements described within.
Release of Funds
To allow Sponsored Projects Administration to release research funds, the PI must document that he/she has IRB approval from both IRBs. To accomplish this, the PI must upload the DoD IRB approval document to the UVM IRB Click approved protocol record as a comment.
Ongoing DoD Oversight
Any substantive modifications to an approved protocol must be reviewed by the UVM as well as the DoD IRB prior to the change in activities.
Scientific Review
The Army and Navy require independent scientific review and approval of nonexempt research prior to
IRB review of new applications and substantive modifications. The review may be conducted by the
funding agency (including DoD), through the use of an established internal review mechanism in the PI’s
school or department, or via an ad hoc scientific review by the researcher’s chair or dean. The review as well as the name and qualifications of the reviewers should be included in submissions to HRPO and UVM IRBs.
Training
UVM human subjects and good clinical practice training through the CITI course is required. The Navy requires that you take additional certification modules. Check with your DoD representative to determine if you need other DoD research-related training. If you have taken DoD-related training in the past, check with your representative to see if it needs to be renewed.
Intent to Benefit - Does Department of Defense allow for a waiver of informed consent?
Funds may not be used for human subject research unless (1) the informed consent of the subject is obtained in advance; or (2) in the case of research intended to be beneficial to the subject, the informed consent may be obtained from a legal representative of the subject.
A waiver of informed consent prior to research activities is prohibited unless the waiver is obtained from the Assistant Secretary of Defense for Research and Engineering ASD(R&E) or a delegated head of the Department of Defense component. This waiver must be submitted as part of the DoD and UVM IRB applications.
This is only applicable to intervention studies.
Surrogate Consent
An individual who is not legally competent to provide consent may not be enrolled unless the research is intended to benefit each subject enrolled in the study, to include subjects enrolled in study placebo arms.
The HRPO and UVM IRBs determine whether there is a benefit to the subject.
Receipt of the DoD IRB approval indicates to UVM that the investigator has completed all the requirements of the DoD.
8.9 Exception from Informed Consent for Emergency Research
New 10/02/20
The IRB may approve an exception to the requirements for informed consent for research on life-threatening conditions for which available treatments are unproven or unsatisfactory and where it is not possible to obtain informed consent from research participants or their legally authorized representatives.
The purpose of this policy is to outline the additional protections required by the regulations for planned emergency research where the requirements for informed consent are waived.
21 CFR 50.24, FDA Guidance “Exception from Informed Consent Requirements for Emergency Research” (04/13) and OHRP Guidance “Informed Consent Requirements in Emergency Research” (OPRR Reports 97-01, 10/31/96) were referenced in development of this policy.
General Information
- Persons with life-threatening conditions who cannot either provide informed consent or refuse research participation are considered to be a vulnerable population. The lack of subject autonomy and inability of subjects to provide informed consent require that additional protections are provided in the review, approval, and performance of this research.
- Prior and continuing IRB reviews are required for planned emergency research. The IRB must approve both the research and the exception to the requirements for informed consent (i.e., waiver) by finding and documenting that the regulatory criteria described below are met.
- To approve a waiver of informed consent for research conducted in emergency settings, a licensed physician who is a member (or consultant) of the IRB and who is not otherwise participating in the research must agree with the IRB’s determination that the criteria for consent waiver are met. Documentation of the physician’s concurrence is also required for approval; therefore, IRB meeting minutes will specifically record the physician’s vote when planned emergency research is reviewed.
- If an emergency research study is subject to both FDA and HHS regulations, the study may not involve prisoners or fetuses, and the provisions of FDA 21 CFR 50.24 must be satisfied.
Definitions
Planned Emergency Research: Research involving human subjects who are in need of emergency medical intervention, but who cannot give informed consent because of their life-threatening medical conditions and who do not have an available legally authorized representative to provide consent.
Emergency Use: Planned emergency research conducted in life-threatening situations must be differentiated from the “emergency use” of an investigational drug or biologic or unapproved medical device. The emergency use provision in FDA regulations allows for a single use of an investigational drug or biologic or unapproved medical device for a human subject in a life-threatening situation for which no standard acceptable treatment is available and when there is not sufficient time to obtain IRB approval.
Family Member: For purposes of the waiver of informed consent for emergency research, any one of the following legally competent persons: spouse, parent, child (including an adopted child), brother, sister, spouse of a brother or sister, and any individual related by blood or affinity whose close association with the subject is the equivalent of a family relationship.
Exception to the Requirements for Informed Consent
The IRB may approve emergency research without requiring that informed consent is obtained from subjects or their legally authorized representatives only if the IRB finds and documents that each of the following requirements under FDA 21 CFR 50.24 has been met:
1) The human subjects are in a life-threatening situation
- Intervention is required before consent from legally authorized representatives is feasible
- Available treatments are unproven or unsatisfactory
- The relative risks and benefits of the proposed intervention are unknown or thought to be equivalent (or better) compared to standard therapy
- The collection of valid scientific evidence (including evidence from randomized, placebo-controlled studies) is necessary to determine the safety and efficacy of the intervention
2) Obtaining informed consent is not feasible because of all of the following
- The subjects will not be able to give their informed consent as a result of their medical condition(s)
- The intervention under investigation must be administered before consent from the subjects' legally authorized representatives is feasible
- There is no reasonable way to prospectively identify the individuals likely to become eligible for participation in the research
3) Participation in the research holds out the prospect of direct benefit to the subjects because of all of the following
- Subjects are facing a life-threatening situation that necessitates intervention
- Appropriate animal and other preclinical studies have been conducted and the information derived from those studies (and related evidence) supports the potential for the intervention to provide a direct benefit to the individual subjects
- Risks associated with the research are reasonable in relation to what is known about the medical condition of the potential class of subjects, the risks and benefits of standard therapy, and what is known about the risks and benefits of the proposed intervention or activity
4) The research could not practicably be carried out without the IRB approval of a waiver of informed consent
- Recruitment of subjects providing informed consent could bias the science, the science is less rigorous as a result of restricting the research to subjects who can provide consent, or the research would be unreasonably delayed by restricting it to consenting subjects
5) The protocol defines the length of the potential therapeutic window based on scientific evidence, and the investigator has committed to attempting to contact a legally authorized representative for each subject within that window of time and, if feasible, to asking the legally authorized representative for consent rather than proceeding without consent
- Investigators will summarize efforts made to contact legally authorized representatives and provide this information to the IRB at the time of continuing review
6) The IRB has reviewed and approved an informed consent process and consent document meeting all the requirements described in regulations and HRPP policies
- The approved informed consent procedures and consent document are to be used with subjects or their legally authorized representatives when feasible
- The IRB has approved procedures and information to be used when providing an opportunity for a family member to object to the subject’s participation (as described below)
7) Additional protections of the rights and welfare of subjects will be provided, including at least
- Consultation (including consultation carried out by the IRB, where appropriate) with representatives of the communities in which the research will be conducted and from which the subjects will be drawn
- Examples: Holding a public meeting in the community from which the subjects will be drawn to discuss the research, conducting a telephone poll, establishing a separate panel of community members, including community consultants to the IRB, and adding unaffiliated members to the IRB who are representative of the community
- The IRB will consider community input when reviewing the research
- Prior to initiation of the research, public disclosure of plans for the research and its risks and expected benefits to the communities in which the research will be conducted and from which the subjects will be drawn
- Public disclosure of sufficient information following completion of the research to apprise the community and researchers of the study, including the demographic characteristics of the research population and its results
- Establishment of an independent data monitoring committee to exercise oversight of the research
- If obtaining informed consent is not feasible (and a legally authorized representative is not reasonably available), the investigator has committed to attempting to contact within the therapeutic window the subject's family member who is not a legally authorized representative, if feasible, and asking whether he/she objects to the subject's participation in the research
- Only one family member must be consulted and agree (or object) to the subject's participation in the research
- If more than one family member is present and family members disagree, the family members must work out the disagreement to enroll the potential subject
- Investigators will summarize efforts made to contact family members and provide this information to the IRB at the time of continuing review.
Note: If a subject is enrolled in the study with waived consent and the subject dies before a legally authorized representative or family member can be contacted, information about the study (as described below) is to be provided to the subject's legally authorized representative or family member, if feasible.
The IRB will approve procedures to inform the subject, the subject’s legally authorized representative (if the subject remains incapacitated), or a family member (if the legally authorized representative is not reasonably available) of the following at the earliest feasible opportunity:
- That the subject was included in the study
- Details of the research and other information contained in the informed consent document
- That the subject’s participation may be discontinued at any time without penalty or loss of benefits to which the subject is otherwise entitled.
Note: If it is a legally authorized representative or family member that is told about the study and the subject's condition improves, the subject is also to be informed as soon as feasible.
Research Subject to FDA Regulations
For planned emergency research subject to FDA regulations, other specific requirements also apply, as described below.
- The IRB will confirm and document that a separate IND or IDE is obtained for use of the investigational drug, biologic, or device to be studied in the research that clearly identifies the protocol as one that may include subjects who are unable to consent. Note: A separate IND or IDE is required even if an IND for the same drug or an IDE for the same device as the one to be studied already exists.
- If the IRB cannot approve the research either because the criteria described above are not met or because of relevant ethical concerns, documentation of the IRB’s findings will be provided in writing to the investigator and sponsor within 14 days.
The sponsor must promptly disclose this information to FDA and to investigators who have been asked to participate in the research or a “substantially equivalent clinical investigation” and to other IRBs that have been or are asked to review this or a substantially equivalent investigation by that sponsor.
Research Subject to DHHS Regulations
The IRB may approve research subject to DHHS regulations only involving an “emergency research consent waiver” if the IRB finds, documents, and reports to OHRP all of the following:
- The research is not subject to FDA regulations
- The DHHS requirements for waiver of informed consent for emergency research (see “Exception to the Requirements for Informed Consent” above) have been met.
8.9.1 Review Flow for Planned Emergency Research
New 10/02/20
Exceptions from Informed Consent (EFIC) protocols require additional review and feedback from an internal advisory panel and the IRB. Approval is a two-step process as illustrated below.
Exceptions from Informed Consent Process for Emergency Research
Step 1. Preparation
The PI needs to prepare the protocol and pull together all the required materials as listed in the blue boxes above. The protocol materials will be submitted through the UVMClick-IRB system. Please note that there are specific requirements for an EFIC protocol. Complete the Smart form and attach the following supporting documents:
- Protocol
- Consent Process and Consent Form
- Supplemental Form “Request for Review of Emergency Research with Waiver of Consent” form, this includes information about community consultation
- IND/IDE
Step 2. Planned Emergency Research Advisory Panel
An Advisory Panel with representation from both UVM and UVMMC will be convened to conduct a pre-review of the protocol and the proposed community consultation plan. See Section 8.9.4 Operations of Exception from Informed Consent (EFIC) for Emergency Research Advisory Panel.
Step 3. IRB Preliminary Approval to Proceed with Community Consultation
The IRB will conduct a preliminary review of the protocol and the community consultation plan to assess if all the requirements have been met and if the plan is appropriate. Any clarifications or modifications will go back to the investigator for resolution following standard procedures. Once approved, the IRB will release a modified approval of just the Community Consultation plan. The PI can then begin the community consultation plan as outlined in the approved protocol materials.
Step 4. PI to Submit Community Feedback to the IRB
Once the community consultation is complete, the PI must provide documented outcomes of the consultation as described in Section 8.7.3 Data Collection Expectations for Exception from Informed Consent for Emergency Research. The results will be reviewed by the IRB and shared with the EFIC Advisory Panel.
Step 5. EFIC Advisory Panel and IRB Final Approval to Proceed with Enrollment
If the Committee and Advisory Panel are satisfied with outcome of the community outreach, the PI will receive a full approval to begin enrollment. The protocol will be categorized as “high-risk” invoking early reporting to the IRB and potential monitoring visits to ensure compliance with consenting processes.
8.9.2 Designing a Plan for Community Consultation and Public Disclosure in Planned Emergency Research
New 10/02/2020
The purpose of this guidance is to provide an outline for drafting a community consultation and public disclosure plan for multi‐site studies that include an Exception From Informed Consent (EFIC). Requirements for EFIC are described in 21 CFR 50.24 (FDA‐regulated studies) and Federal Register, Vol. 61, pp. 51531‐51533 (non‐FDA‐regulated studies). This guidance does not provide a full accounting of the requirements of community consultation and public disclosure.
Investigators must review the FDA’s community consultation and public disclosure guidance for complete information: FDA Guidance, April 2013: Exception from Informed Consent Requirements for Emergency Research.
Investigators at each research site are required to complete community consultation and public disclosure activities prior to enrolling participants into the study. The goals of community consultation are as follows:
- To ensure that all relevant communities have opportunity for input into the IRB’s decision‐making process before initiation of the study
- To present information so that community members understand the proposed investigation, understand its risks and benefits.
- To be sure community members understand that the investigation will take place without informed consent.
The goal of public disclosure prior to initiation of the study is to provide sufficient information to allow a reasonable assumption that the broader community is aware of the plans for the investigation, its risks and expected benefits, and the fact that the study will be conducted without obtaining informed consent from most study subjects.
The goal of public disclosure after the study is completed is to ensure that the communities, the public, and scientific researchers are aware of the study’s results.
Requirements for the Study Team
It is the responsibility of the study team to design a protocol‐level community consultation and public disclosure plan that can be used throughout the UVMMC’s service region. The plan must take into account the nature of the participant population overall as well as primary differences in the community and resources at the participating locations. Though the study team is responsible for executing the community consultation and public disclosure plan throughout the UVMMC’s service region.
The protocol‐level study plan must include a variety of passive and interactive consultation and disclosure methods and study‐specific supporting materials. The plan must include specific justification for how each method can appropriately notify and solicit feedback from the participant population and the community.
Interactive methods may include the following:
- Standing meetings, such as local civic public forums, may be better attended because such meetings are already on community members' calendars.
- Public community meetings or other special meetings specifically organized to discuss the research. Such meetings may be valuable in attracting participation from individuals with strong interest in the research, e.g. patient support groups, clinicians, IRB members, etc.
- Local radio and/or television talk shows. Such programs allow viewers to "call in" to express their views and concerns.
- Interactive websites, social media, focus groups, and surveys.
Passive methods may include the following:
- Targeted mailings to households in the communities, with information about how to obtain further details.
- Advertisements and articles in the English language, and if appropriate, foreign language, newspapers (Public outreach documents should be translated into languages that are common in the area served by the facility where the investigation is being conducted and in the communities from which subjects will be drawn).
- Clearly marked links and information on the sponsor’s and participating hospitals’ Internet web sites.
- Summary materials that are accessible to non‐English speaking or homeless populations who reside in the community from which research subjects are likely to be drawn.
- Presentation or distribution of information at meetings of community, local government, civic, or patient advocacy groups.
- Letters to local and regional community leaders and first responders (e.g., police, paramedics).
- Announcements to local/regional hospital staff(s).
- Public service announcements and interviews or discussions on “talk” radio or television programs.
- Press conferences and briefings.
- Meetings or activities provided by hospitals’ and institutions’ existing community outreach programs.
The plan must also describe the general content that will be presented during the community consultation activities. Study‐specific materials developed for community consultation should reflect this general content as well. General content should including the following information:
- A summary of the research protocol, study design, and a description of the procedures to be followed, including the identification of any procedures which are experimental.
- A summary of other available treatment options and what is known about their risks and benefits.
- An estimate of how long the study will last and expected duration of the subject’s participation.
- How potential study subjects will be identified.
- Information about the test article’s use, including a balanced description of the risks and expected benefits and any relevant information that is known about adverse events.
- A clear statement that prospective informed consent will not be obtained for most research subjects.
- The rationale as to why the study must be conducted using an exception from informed consent.
- A copy of the informed consent document.
- Relevant information that would be part of the informed consent process (21 CFR 50.25(a) and (b), as applicable), e.g., available treatments for the condition under study; risks/potential benefits of participating in the research; possibility that FDA might inspect the subject’s records.
- A description of the therapeutic window, during which the test article must administered, and the portion of that window that will be used to contact the subject's LAR.
- A description of the attempts that will be made to contact the subject's LAR to obtain consent, or, if no LAR is available, a family member to provide an opportunity to object to the subject’s enrollment in the study, both before and after the test article is administered.
- A description of the way(s) in which an individual may express his/her desire not to participate and avoid involvement as a subject in the research (e.g., opt‐out mechanisms), if any will be made available.
- Reasons why community input is important.
- Known community perceptions/concerns associated with the study, product, and/or standard of care.
- Identification of individuals to contact for more information about the study.
Requirements for the Study Team
It is the responsibility of the study team to use the protocol‐level community consultation and public disclosure plan to design and implement a site‐specific plan. The study teams must select the passive and interactive consultation and disclosure methods that are most appropriate and feasible for implementation throughout the UVMMC service region. The plan must describe how the selected methods will be executed and justification for how each method can appropriately notify and solicit feedback from the participant population and the community.
The plan must address the needs of the participant population and community, which many include the following:
- Cultural, demographic, geographic, and economic considerations
- Languages and local educational and/or literacy concerns
- Religious, social, and political considerations
In addition to the written plan, the study team must complete the Site Community Profile Worksheet, which will help the study team identify and describe the composition of the community.
In preparing to execute the plan, the study team must also be prepared to collect data regarding the results and feedback provided through community consultation and disclosure methods. The plan must include a description of how the study team will collect and report on this data.
8.9.3 Data Collection Expectations for Exception from Informed Consent for Emergency Research
New 10/02/20
When determining whether the community consultation and disclosure process is adequate for an EFIC study, the IRB must consider the community's opinions and concerns, assess the adequacy of the consultation and disclosure process, and incorporate the results of community consultation and discussion into its decision making.
The IRB may wish to directly hear the community discussions and concerns expressed in those discussions, and not rely solely on summary documentation by the clinical investigator or feedback reported by others, so it is recommended that community discussions be recorded in some way, and that community members be informed that the minutes and/or audio/video recordings of discussions may be reviewed by the IRB.
The study team conducting consultation and disclosure procedures must provide the IRB with the results of the process in a Community Consultation and Disclosure Report. Quantitative results are helpful, but qualitative information is also requested.
Data Collection Expectations
Interactive Consultation
For example, live events, standing meetings such as local civic public forums, public community meetings or other special meetings specifically organized to discuss the research, focus groups and surveys, and local radio and/or television talk shows:
- Date, time, and location of event, if applicable
- Information presented by the study team and the length of the presentation
- Number of community members in attendance
- Responses to survey/focus group questions, if applicable
- Amount of time allotted for community questions and feedback
- Questions or concerns raised by community members (grouped by common themes), if applicable;
How were questions or concerns from the audience collected? How were questions or concerns from the audience addressed? What were the outcomes of these meetings?
Passive Methods
For example, mailings, websites, fliers, letters, announcements, press conference briefings, advertisements, newsletters, etc.:
- Date information was made public
- Location(s) the information was posted or sent
- Questions or concerns raised by community members (grouped by common themes), if applicable;
How were questions or concerns from the audience collected? How were questions or concerns from the audience addressed? What were the outcomes of these discussions?
8.9.4 Operations of Exception from Informed Consent for Emergency Research Advisory Panel
New 10/02/20
The Exception from Informed Consent for Emergency Research Advisory Panel (EFIC) is a group of stakeholders across UVM and UVMMC that will convene to review planned emergency research protocols. Planned Emergency research is research that is conducted without prior informed consent from research subjects, see Section 8.7. The EFIC Panel’s role is advisory in nature. The panel will confirm, prior to the IRB review, that the project is suitable for UVMMC and that they agree the proposed community outreach plan is acceptable in light of the vulnerability of the intended subjects.
Composition of the EFIC Advisory Panel
Representation from each of the institutions is as follows:
UVMMC Chief Medical Officer
College Dean
VP for Research
Office of Legal Counsel (UVMMC/UVM)
Office of Communications (UVMMC/UVM)
Ethics (UVMMC)
Office of Governmental Relations (UVMMC/UVM)
Patient and Family Advisory Committee (UVMMC)
Risk Management (UVM)
Emergency Department (UVMMC)
Emergency Medical Services
Advisory Panel Review of EFIC Protocols
Review of these protocols with undergo the following procedures.
Initial Review of Protocol
Electronic Review
Protocol materials will be disseminated to the EFIC Advisory panel members through the UVMClick-IRB ancillary review procedure. Members receive an email notice that an ancillary review is pending. Members are required to authenticate into the electronic system using their UVM NetID and password prior to completing their review. The system validates the member’s authentication credentials based upon the member’s role in the system and determines available actions for each person. The electronic review is stamped within the system with the name of the individual carrying out the review activity (electronic signature), and the time and date that the electronic signature was applied to the review.
Convened Meeting
The EFIC Advisory Panel is convened after all electronic reviews are completed within the UVMClick-IRB system.
Voting Requirements
- A majority of the total number of regular voting members will constitute a quorum. The number in attendance must be one more than half the total number of regular voting members. If less than a majority of the total number of regular voting members is present, one or more alternate members may be included to constitute a quorum if they have been specifically designated to alternate for a specific absent member.
- At least one member from the UVMMC Patient & Family Advisory Committee must be present to constitute a quorum.
- At least one IRB community member must be present to constitute a quorum.
- Official panel decisions will be by formal vote of a simple majority at convened meetings of a quorum of members.
Outcomes of Initial EFIC Panel Review
- Approval to move forward with IRB submission, no issues.
- Approval to move forward with minor clarifications, can be signed off by IRB through their review.
- Substantive issues/clarifications to the PI prior to IRB review.
- Determination that study will not be carried out by the institutions.
Review of Community Feedback
Electronic Review
A summary of the community feedback will be disseminated to the EFIC Advisory panel members through the UVMClick-IRB ancillary review procedure. All responses will be collated and presented to the Chief Medical Officer for final approval to move forward with study enrollment.
Outcomes of EFIC Panel Review of Community Feedback
- Approval to move forward with IRB final approval and enrollment.
- Modifications required to address negative or insufficient feedback from the community.
Signing Authority to Recommend Protocol be Submitted to the IRB and to Allow Subject Enrollment
The Chief Medical Officer, with input from the EFIC Advisory Panel, will approve the protocol to be reviewed by the IRB as well as approve enrollment after Panel review of the community feedback.
Documentation of Review
The IRB will maintain all reviews within the UVMClick-IRB protocol record and minutes of meetings will be scanned and uploaded to the system.
The member roster will be maintained within the UVMClick-IRB module. Rosters are updated each time there is a change in the membership.
8.10 Research Conducted in Public Schools
New 08/30/2022
Federal regulations consider children to be vulnerable populations. Investigators proposing to conduct research in public elementary and secondary schools must review this guidance as well as review Sections 23.4 and 9.1, which address children in research and the appropriate methods of consenting/assenting children to participate in research. Please note some of this guidance would also apply to many freshmen in college (17 years old or younger).
Conducting Research in an Educational Institution
Researchers must obtain a letter signed by the school Principal or district superintendent or other individual with authority to commit the school’s resources, acknowledging the proposed research activity, and granting permission for the engagement of their students, employee(s) and facilities (if applicable) in that activity. A template of our support letter can be found here.
Accessing Educational Records - Family Educational Rights and Privacy Act (FERPA)
If a researcher wants to access the educational records of students, the Family Educational Rights and Privacy Act (FERPA) applies. FERPA is a Federal law administered by the U.S. Department of Education; 34 CFR Part 99, which protects the privacy of student education records.
Applicability and Requirements
FERPA applies to all educational agencies and institutions that receive federal funding. For information regarding FERPA at UVM, please see FERPA Rights Disclosure on the UVM Policy page.
Education records include any record containing personally identifiable information (PII) directly related to the student. PII is not limited to name but may include indirect identifiers as well. FERPA stipulates that educational institutions have the authority to determine what information is accessed from an education record. If an institution denies an investigator access to information in an education record, the IRB cannot overrule the decision. Examples include:
Documents with a student’s name, ID number, or other identifier;
- Class rosters or grade lists;
- Place of birth;
- Ethnicity;
- Residency status;
- Advisor’s name;
- Class schedule;
- Courses completed;
- Grades;
- Disciplinary records;
- Student info displayed on a computer screen.
Student education records are accessible to and used by instructors, teachers, and administrators for the purposes of conducting the duties of their job. For example, as part of a teacher’s job, there is natural access to student’s assignments, test scores, and attendance records in order to evaluate performance and ultimately assign a grade. However, this same teacher cannot use this natural access for other intents and purposes, such as research. If a teacher/instructor wants to use student data for research purposes, FERPA applies and consent is required, unless one of the exceptions to consent as outlined below is met.
Consent Requirements under FERPA to Access Educational Records
Use of educational records for research purposes requires parental permission and assent from the student as applicable, see section 9.1. Consent form must:
- Specify the records to be disclosed;
- State the purpose of the disclosure;
- Identify the party to whom the disclosure is to be made;
- Include a dated student signature. This may include a signature in an electronic form that:
1) Identifies and authenticates a particular person as the source of the electronic consent; and
2) Indicates such a person's approval of the information contained in the electronic consent.
Consent Requirement Exceptions under FERPA to Access Educational Records
A list of exceptions allowing for the use of educational records for research purposes without consent is below. If any of these situations apply, please include any applicable agreements in your IRB submission.
- The only PII obtained constitutes “directory information” and the student has not opted out of having his/her information included in the directory. However, schools must tell parents and eligible students that directory information is not protected, and they must allow parents and eligible students a reasonable amount of time to request that the school not disclose directory information about them. At the elementary and middle school levels, often times this is something parents agree to at the beginning of each school year.
- At UVM, the Registrar’s Office maintains the list of students who have opted out of the directory;
- The release is to an authorized representative of state/local educational authorities for an audit or evaluation of federal or state supported education programs, or for the enforcement of or compliance with federal legal requirements related to those programs;
- Investigators must provide IRB with evidence that they are acting as authorized representatives of a state or local educational authority and that their audit or evaluation meets the conditions described above (e.g. a Letter of Intent, Memorandum of Understanding between university and educational authority);
- The release is to organizations conducting studies for or on behalf of educational agencies or institutions to develop, validate or administer predictive tests; administer student aid programs; or improve instruction;
- A written agreement, which meets criteria listed in FERPA between the university and the educational agency or institution is required.
When Research is funded by the U.S. Department of Education
There are additional protections for students when the research is funded by the Department of Education. The Protection of Pupil Rights Amendment (PPRA), also known as “Students Rights in Research, Experimental Programs and Teaching (20 U.S.C. § 1232h; 34 CFR Part 98) is a federal law that affords certain rights to parents of minor students with regard to surveys that ask questions of a personal nature. The No Child Left Behind Act of 2001 contains a major amendment to PPRA that gives parents more rights with regard to the surveying of minor students, the collection of information from students for marketing purposes, and certain non-emergency medical examinations.
Applicability and Requirements of PPRA
The PPRA regulation applies when the Department of Education funding is either (1) direct funding of a particular research topic by the department, or (2) general school funding from the department.
The law requires that schools obtain written consent from parents before minor students participate in any U.S. Department of Education funded survey, analysis, or evaluation that reveals information concerning the following areas:
- Political affiliations;
- Mental and psychological problems potentially embarrassing to the student and his/her family;
- Sex behavior and attitudes;
- Illegal, anti-social, self-incriminating and demeaning behavior;
- Critical appraisals of other individuals with whom respondents have close family relationships;
- Legally recognized privileged or analogous relationships, such as those of lawyers, physicians, and ministers;
- Religious practices, affiliations, or beliefs of the student or student's parent*; or
- Income (other than that required by law to determine eligibility for participation in a program or for receiving financial assistance under such program.)
Note: Parental permission for the students to participate cannot be waived.
PPRA also addresses marketing surveys and other areas of student privacy, parental access to information, and the administration of certain physical examinations to minors. The rights under PPRA transfer from the parents to a student who is 18 years old or an emancipated minor under state law.
FERPA and PPRA in Private Schools
A private school that does not receive any federal funding is not subject to the provisions of FERPA or PPRA. However, if research in a private school is directly funded by the Department of Education, PPRA applies.
Other Considerations
Informed Consent (Assent, Parental Permission)
As with other research, obtaining informed consent is required for research conducted in schools. Generally, the IRB requires assent for school-aged students (ages 11-17). Parental permission is also required for children to participate in research. Obtaining parental permission and assent in school-based research may present challenges to the investigator. Investigators may want to work closely with school administrators or teachers as their support may be important in the return of consent forms and/or study materials. A thorough description of how researchers will obtain parental permission and assent must be included in the protocol submission and approved by the IRB prior to initiation.
Plans to Avoid Coercion
Care should be taken so that children do not feel pressured to participate in research. When research is conducted in the schools, younger children may need to be reassured that their teacher will not be mad at them if they do not want to take part in the research activities. Alternative activities should be made available that do not single out children who choose not to take part. Adolescents who might be vulnerable to peer pressure may need privacy to make their decisions. Depending on the age of the children, it may be appropriate to tell them that there are no right or wrong answers to the researcher's questions. Efforts to protect children from undue pressure must be included in the protocol submission.
8.11 International Research: Information on Conducting Research Outside of the United States
Revised: 3/15/22
Research conducted at the expedited or full review level by University of Vermont faculty, staff or students in foreign countries poses unique and complex ethical challenges. Each country has different cultures and values, and it is crucial to understand the local context. As a result, the IRB expects you to acknowledge and understand the following:
- Researchers must obtain IRB approval before the study can begin. All faculty members, staff or students, must have IRB approval before initiating research in a foreign country.
- Demonstrate cultural understanding and sensitivity. Protocols should describe any anticipated cultural sensitivities of conducting the research and how researchers intend to address these sensitivities. The researcher should be familiar with local customs, culture, and religious norms in the country where the study will be conducted. Is the typical process of signing an informed consent document culturally acceptable for your study? How should recruitment be done? Are there other cultural issues you might encounter once you arrive? If field work is proposed, is a verbal consent process and documentation needed?
- Understand the research ethics guidelines of the host country. Investigators will be required to obtain IRB approval for research done internationally from both the UVM IRB and the local IRB/Ethics Committee within the host country where the research will be conducted. The UVM IRB requires the host country IRB/Ethics approval be on file with UVM’s IRB prior to UVM IRB approval being granted. The IRB strongly recommends you clearly understand the host country's requirements for reviewing and approving human participant research. Some countries have clear ethical guidelines that must be met for conducting domestic and/or international research. Other countries will not have a formal process but might rely on other neighboring countries to assist with the review.
What if there is no local IRB review in the country? If there is no local IRB that will be reviewing the study, then the UVM IRB will require some additional information to enable an analysis of risk to benefit for those participants. This should take the form of a letter from an authoritative source in that host country, an advisor, advisory board member, dissertation committee member, or a UVM faculty member. The author of this document must be provided with a summary of the intended research and the anticipated procedures and processes. The author must be someone who is familiar with the culture, customs and norms of the setting and be able to vouch for the appropriateness of the activities and/or consenting process as well as the ability of the research team to carry out all study activities. The signed letter must include the following information
- how the author has expertise in the culture and social norms of the country/region where the study will take place;
- that the individual providing this review has been provided with a summary of the research and the proposed procedures;
- Provision of resolutions to challenges in the conduct of the research in a respectful and safe manner; and,
- an affirmation that the research is appropriate.
International Research Standard Resources
The Office of Human Research Protections (OHRP) publishes the International Compilation of Human Research Standards, is a listing of over 1,000 laws, regulations, and guidelines on human subjects protections in 133 countries and from many international organizations. Most of the listings provide hyperlinks to the source document. These laws, regulations, and guidelines are classified into nine categories:. Researchers should check this document to determine the countries applicable laws, regulations and guidelines on Human Participants Researcher.
- The Office of Human Research Protection (OHRP) has issued a Listing of 27 Social-Behavioral Research Standards . This includes laws, guidelines, and regulations applicable to social-behavioral research around the world. The standards were developed in 18 countries and by one international organization. The standards are organized by continent, and then arranged alphabetically by country name. Researchers are encouraged to review table 1 and determine if their country of research has any specific restrictions or standards related to international research.
- Know the data laws. While not specifically under the IRB’s domain, you should know that there are some restrictions on bringing identifiable data into/out of some countries. The EU, for example, has laws surrounding what kind of identifiable information can be taken out of Europe and brought to the US (this applies to electronic data that will be housed on a US server as well). Please refer to section 10.3 European Union (EU) Participants and EU General Data Protections (GDPR)
IRB Review
The IRB relies on the information you provide to help assess whether the right protections are in place for participants. In addition to the usually required information submitted for review to the IRB, the following points should be addressed in your submission.
- Your IRB protocol should describe relevant local context information, any anticipated cultural sensitivities of conducting your research and how you intend to overcome identified issues. This should include, but not limited to the following:
- Cities, regions countries where research will be conducted
- Scientific/ethical justification for conducting the research in an international setting
- Current events or socio-political environment in the country that may impact research conduct or alter the risks or benefits to participants
- Societal and cultural beliefs in the country that may impact research conduct or alter the risks or benefits to participants
- Literacy rate of the potential participant population
- Languages and dialects of the potential participant population
- Description of the research team’s knowledge of or experience in the host country
- Distribution of risks and current and future benefits
- Approval letter from the local IRB/Ethics Committee, equivalent board, or group within the country where research will be conducted. If there is no such oversight mechanism, researchers must provide letter as explained above.
- The consent form should be submitted in both the local language of the host country and in English. Please clearly label each form for the IRB. The application should also indicate who conducted the translation of the forms and provide a letter certifying the translations are correct.
- Recruitment materials to be used in both the local language of the host country and in English.
- Describe how you will keep your data secure at all stages: while you are collecting it in the host country, while you are traveling back to the US and once you arrive here.
9. Consent (§____.116)
Revised 10/02/10
Legally Effective and Prospectively Obtained Informed Consent and Documentation of Consent
Informed Consent
Informed Consent is an individual’s voluntary agreement, based upon adequate knowledge and understanding of relevant information, to participate in research or to undergo a diagnostic, therapeutic, or preventive procedure.
Informed consent must be obtained from the participant or their legally authorized representative using most recently-approved version of the consent document prior to initiating research activities.
IRB Review of Consent
The IRB ensures that provisions are made to obtain legally effective informed consent prospectively from each prospective research participant or permission from his/her legally authorized representative, in accordance with and to the extent required by 45 CFR 46.116 and 21 CFR 50.20. However, there are circumstances in which the IRB may grant a waiver of informed consent in accordance with Federal regulations. (see section titled Waiver of Informed Consent, Alteration of Informed Consent, or Waiver of Documentation for more information.)
Written informed consent is to be obtained unless alternate procedures are approved by the IRB, in accordance with 45 CFR 46.117 and 21 CFR 50.27. The IRB reviews all informed consent documents to assure the adequacy of the information contained in the consent document, and adherence to Federal regulations regarding the required elements of informed consent.
Consent Requirements/Elements in the Form
An investigator may not involve a human participant in research without first obtaining the informed consent (with HIPAA authorization language included when Protected Health Information (PHI) is used/disclosed) of the participant or the participant's legally authorized representative. This must be obtained in writing, signed electronically (a written copy must be provided to the participant)(46.117(a)) or verbally (if the specific criteria as described below are met). In either case, informed consent (and HIPAA, when applicable) must be obtained under circumstances which allow sufficient opportunity to consider participation and that minimize the possibility of any coercion or undue influence.
The information that is given to participants must be in a language understandable to them or their representative. Whether informed consent is written or oral, it must not include any exculpatory language through which the participant or representative is made to waive or appear to waive any of the participant's legal rights, or releases or appears to release the investigator, sponsor, institution or its agents from liability for negligence.
If genetic materials are to be collected, stored or analyzed, GINA language must be included in the participant consent form. Language can be found in the IRB consent template located on our forms page.
As of January 2018 consent forms should contain the following:
- A concise summary of study activities, risks, and benefits presented to research participants in advance of the body of the consent document. The IRB will not require re-consent for already enrolled participants.
The IRB provides instructions and a consent template to assist with consent form development. The elements are listed below.
Basic Elements of Informed Consent: §____.116(a)
- A statement that the study involves research, an explanation of the purposes of the research and the expected duration of the participant's participation, a description of the procedures to be followed, and identification of any procedures which are experimental.
- A description of any reasonably foreseeable risks (physical, psychological, social, legal, or others) or discomforts to the participant.
NOTE: See section titled Elements Found in a Standard Clinical Trial Protocol for additional information regarding GINA. - A description of any benefits to the participant or to others, which may reasonably be expected from the research. If there is no direct benefit to the participant, this should be stated.
- A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the participant.
- A statement describing the extent to which confidentiality of records will be maintained.
- For research involving more than minimal risk, an explanation as to whether compensation and any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained. When applicable, standard language from the template must be used as written.
- An explanation of whom to contact for answers to pertinent questions about the research and research participants' rights, and whom to contact in the event of a research-related injury to the participant.
- A statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the participant is otherwise entitled and the participants may discontinue participation at any time without penalty or loss of benefits.
- The name, address, and telephone number of the principal investigator(s) or contact person(s).
- The amount of compensation, if any, for participation.
- Identifiers might be removed and the de-identified information or biospecimens used for future research without additional informed consent from the participant; or the participant’s information or biospecimens will not be used or distributed for future research studies even if identifiers are removed.
Additional Elements of Informed Consent: §____.116(b)
- A statement that the particular treatment or procedure may involve risks to the participant (or to the embryo or fetus, if the participant is or may become pregnant) which are currently unforeseeable.
- Anticipated circumstances under which the participant's participation may be terminated by the investigator without regard to the participant's consent.
- Any additional costs to the participant that may result from participation in the research.
- The consequences of a participant's decision to withdraw from the research and procedures for orderly termination of participation by the participant.
- A statement that significant new findings developed during the course of the research which may relate to the participant's willingness to continue participation will be provided to the participant.
- The approximate number of participants involved in the study.
- When HIV testing is conducted as part of the research procedures, individuals whose test results are associated with personal identifiers must be informed that they will be provided with their test results and provided with the opportunity to receive appropriate counseling. It should also be stated that both HIV and AIDS cases must, by law, be reported to the Vermont Department of Health and disclosure of a positive test may result in discrimination by friends, family, employers, insurance companies and others. See consent template for additional guidance.
- 46.116(c)(7) - A statement that the participant’s biospecimens (even if identifiers are removed) may be used for commercial profit and whether the participant will or will not share in this commercial profit.
- 46.116(c)(8) - A statement regarding whether clinically relevant research results, including individual research results, will be disclosed to participants, and if so, under what conditions.
- 46.116(c)(9) - For research involving biospecimens, whether the research will (if known) or might include whole genome sequencing (i.e., sequencing of a human germline or somatic specimen with the intent to generate the genome or exome sequence of that specimen).
For consent form document guidance as well as templates for various types of consents, see the Consent and HIPAA Guidance page of the IRB website.
Flexibility in Obtaining Consent
The process that will be used to obtain consent must be outlined in the protocol submission. The consent process may be in person or remote, it may require a witness, or a legally authorized representative. Each of these different scenarios have guidance within the applicable sections of the manual.
Regulations allow flexibility in the way written consent is obtained. In addition to pen and paper consent, technology now allows for written consent to be obtained electronically.
Obtaining Electronic Written Consent
New technology now allows investigators to obtain written consent electronically. See Section 9.9 Obtaining Electronic Written Consent for more information.
Obtaining Written Consent using Email or Fax
Investigators are allowed to exchange consent documents for signature via email and/or fax following the steps outlined.
1. An investigator and/or designee uses email/fax to send an unsigned copy of the current IRB-approved version of the ICF for participant signature. To protect participants’ privacy, researchers must encrypt email referencing participation in research, including the consent form. Information about how to encrypt email can be found here.
2. The potential participant is contacted via phone or videoconferencing to review the consent document and discuss participation. When conducting the consent discussion first verify the participant’s/legally authorized representative’s identity, then verify that:
- The form the participant received is the currently approved version;
- That all the pages of the consent were received; and
- That the participant is able to read all the pages of the consent.
Once verified, review all elements of the consent with the participant and pose them questions to demonstrate their understanding of participation in the research study.
3. Once all of the participant’s questions have been answered, the participant signs the consent form. There are multiple options for the participant to return the signed copy. Researchers must provide technical assistance to participants when needed.
- US Mail the signed copy back to researcher;
- If participant has a printer/scanner, scan the signed copy and email back to the investigator.
- They can take a picture of the signature page and either email or text that back to the investigator.
Note: Regulations require that participants be provided with a copy of the consent document. In this instance, because the consent was sent to them via email or fax, this requirement is met.
Document this information and the consent process using one of the consent process documentation examples. The template should be modified to capture all of the above information (e.g. how contacted, how identity ascertained, date/time, etc).
4. Investigators will receive and complete their signature and the current date on the partially executed consent form. Investigators need to maintain the fully executed copy of the entire consent form along with the consent process documentation. This is Good Clinical practice and may be reviewed as part of a quality assurance visit.
Documentation of Consent
The process of administering a consent for research must be documented. This requirement is based on 21 CFR 312.62(b) and 812.150(a)(3)(i) which states that “The case history for each individual shall document that informed consent was obtained prior to participation in the study. “ The approaches to verify consent documentation are flexible. Documentation may be contained in a case report form, participant’s individual medical record, (e.g. progress notes of the physician, on the participant’s hospital chart, in nursing notes), SMART phrases in the EMR, or consent documentation templates available on the IRB website.
When a research study is inspected/audited, the inspectors will be looking for evidence that consent was obtained prior to any research activities and thus having administration of the consent documentation, as described above, will fulfill this requirement.
Posting of Clinical Trial Consent Form (New Information)
46.116(h)(1-3) There will be new requirements for posting clinical trial consent forms on a publicly available federal website that will be established as a repository for clinical trial consent forms.
(1) The responsibility for posting is on the awardee or the federal department or agency component conducting the study. The posting can take place any time after the trial is closed to recruitment, so long as the posting is no later than 60 days after the last study visit by any participant (as required by the protocol).
(2) The redaction of proprietary or institutionally sensitive information of portions of the consent forms is allowed.
(3) Only one version (no necessarily the final) of the consent form (absent any signatures) for each clinical trial must be posted on the federal website after the clinical trial is closed to recruitment. In accord with the new single IRB review requirement, only one posting is required for each multi-institution study. There is no expectation that a version would need to be posted for each study site nor even for each class of participants in the study (for example, a posting both for adults and for children).
For more information regarding posting informed consents go here https://www.hhs.gov/ohrp/regulations-and-policy/informed-consent-posting/index.html
9.1 Children
Revised 2/6/2023
For any protocol involving children, the IRB must determine which of four categories (see categories below) of research apply to that study, if any. The IRB will document the rationale for this choice in the minutes.
For subjects less than 18 years of age, their parent(s) or other legal guardians are the legally authorized representatives who may grant permission for their participation in research (in accordance with 45 CFR 46 (children’s subpart D).
Definitions:
- Assent: An individual’s affirmative agreement to participate in research obtained in conjunction with permission from the individual’s parents or legally authorized representative. Mere failure to object should not, absent affirmative agreement, be construed as assent. The default age for written assent at UVM/UVM MC is age 11 so if all subjects will be younger than 11 years, then the requirement for written assent may be waived.
- Children: Any person who has not attained 18 years of age.
The HHS regulations at 45 CFR part 46, subpart D permit IRBs to approve the following categories of research involving children as subjects:
- Pediatric Risk Level 1 - Research not involving greater than minimal risk to the children 45 CFR 46.404
To approve this category of research, the IRB must make the following determinations:
• the research presents no greater than minimal risk to the children; and
• adequate provisions are made for soliciting the assent of the children and the permission of their parents or guardians, as set forth in HHS regulations at 45 CFR 46.408. - Pediatric Risk Level II - Research involving greater than minimal risk but presenting the prospect of direct benefit to the individual child subjects involved in the research 45 CFR 46.405
To approve research in this category, the IRB must make the following determinations:
• the risk is justified by the anticipated benefits to the subjects;
• the relation of the anticipated benefit to the risk presented by the study is at least as favorable to the subjects as that provided by available alternative approaches; and
• adequate provisions are made for soliciting the assent of the children and the permission of their parents or guardians, as set forth in HHS regulations at 45 CFR 46.408. - Pediatric Risk Level III - Research involving greater than minimal risk and no prospect of direct benefit to the individual child subjects involved in the research, but likely to yield generalizable knowledge about the subject's disorder or condition 45 CFR 46.406
In order to approve research in this category, the IRB must make the following determinations:
• the risk of the research represents a minor increase over minimal risk;
• the intervention or procedure presents experiences to the child subjects that are reasonably commensurate with those inherent in their actual, or expected medical, dental, psychological, social, or educational situations;
• the intervention or procedure is likely to yield generalizable knowledge about the subject's disorder or condition which is of vital importance for the understanding or amelioration of the disorder or condition; and
• adequate provisions are made for soliciting the assent of the children and the permission of their parents or guardians, as set forth in HHS regulations at 45 CFR 46.408.
A fourth category of research requires a special level of HHS review beyond that provided by the IRB.
- Pediatric Risk Level IV - Research that the IRB believes does not meet the conditions of 45 CFR 46.404, 46.405, or 46.406, but finds that the research presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of children 45 CFR 46.407
If the IRB believes that the research does not meet the requirements of 45 CFR 46.404, 46.405, or 46.406, but finds that it presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of children, it may refer the protocol to HHS for review. The research may proceed only if the Secretary, HHS, or his or her designee, after consulting with a panel of experts in pertinent disciplines (e.g., science, medicine, education, ethics, law) and following an opportunity for public review and comment, determines either: (1) that the research in fact satisfies the conditions of 45 CFR 46.404, 46.405, or 46.406, or (2) the following:
• the research presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of children;
• the research will be conducted in accordance with sound ethical principles; and
• adequate provisions are made for soliciting the assent of children and the permission of their parents or guardians, as set forth in HHS regulations at 45 CFR 46.408.
For more information on the HHS 45 CFR 46.407 review process see OHRP Guidance, Children Involved as Subjects in Research: Guidance on the HHS 45 CFR 46.407 ("407") Review Process
Consenting Children
The consent process of children should meet the following Requirements for Assent and Parental Permission:
Pediatric Risk Level I - Research not involving greater than minimal risk to the children 45 CFR 46.404 The UVM IRB has determined that the written assent of all children between age 11-17 must be sought and documented unless the protocol has applied and been granted a waiver. Verbal assent should also be documented for children under age 11 if the child is capable of providing assent accounting for the ages, maturity, and psychological state of the children involved.
Pediatric Risk Level II - Research involving greater than minimal risk but presenting the prospect of direct benefit to the individual subjects 45 CFR 405, Adequate provisions are made for soliciting the assent of the children and permission of at least one parent or guardian, as set forth in 46.408. The UVM IRB has determined that the written assent of all children between age 11-17 must be sought and documented and a verbal assent be documented for children under age 11 if the child is capable of providing assent accounting for the ages, maturity, and psychological state of the children involved.
Pediatric Risk Level III and IV- Permission must be obtained from both parents, unless one parent is deceased, unknown, incompetent or not reasonably available, or when only one parent has legal responsibility for the care and custody of the child.
If researchers determine one parent is “not reasonably available” and would like to enroll a child with only one parent consenting, researchers should contact their IRB regulatory analyst to discuss the criteria prior to enrolling. “Not reasonably available” does not apply to situations when a parent is at work, traveling, not immediately available by electronic means, or living in another state or country, without more to justify the investigator’s inability to reach the parent and seek permission.
Examples of situations when one may reasonably conclude that a parent is not reasonably available could include the following situations:
- The parent is incarcerated and not contactable.
- The parent is on active military duty and not contactable.
- The parent’s whereabouts are unknown.
- The parent is known and contactable but chooses not to be involved in the child’s care.
- The parent is known but, upon inquiry, there is reason to believe that requesting permission would be inconsistent with the parent/child relationship, such as where there is reason to believe there is or has been domestic violence or other situations involving harm to the health or welfare of the child.
After consultation with an IRB Chair, situations may be approved in rare circumstances. Please refer to the guidance from OHRP focusing on Parental Permission in Research involving Children.
UVM IRB policy considerations on child dissent in research
The Committee also agrees that dissent of a child (i.e., their actual objection to research) should be considered binding in non-therapeutic, pediatric risk level I research. If a child is capable of assent and the Institutional Review Board (IRB) requires that assent be sought, it must be obtained before the child can participate in the research activity.
***The assent of a child is not a necessary condition for proceeding with the research if the IRB finds that the capability of some or all of the children is so limited that they cannot be reasonably consulted or that the intervention or procedure involved in the research holds out a prospect of direct benefit that is important to the health or well-being of the children and is available only in the context of the research. Permission of the parents or legally authorized representative is still a federal requirement.
Parents
Only the parent(s) may grant permission for the child’s participation in research. Assent is to be sought from the child, only after permission has been obtained from one or both of the parents.
Grandparents and other relatives or caregivers may not grant permission unless they have been granted formal custody of the child by a court. In such cases, the Principal Investigator (PI) must obtain a copy of the court order as evidence of that person’s authority to grant permission for participation in research on the child’s behalf. See additional information regarding consent process, see below.
Waiver of Consent (and HIPAA Authorization if PHI is involved)
If the IRB determines that a research protocol is designed for conditions or for a subject population, for which parental or guardian permission is not a reasonable requirement to protect the subjects (for example, neglected or abused children), the IRB may waive the consent requirements, provided an appropriate mechanism for protecting the children who will participate as subjects in the research is substituted, and provided further that the waiver is not inconsistent with federal, state or local law. The choice of an appropriate mechanism would depend upon the nature and purpose of the activities described in the protocol, the risks and anticipated benefits to the research subjects, age, maturity, and condition.
NOTE: The FDA does not typically allow for a waiver of consent when regulated test articles are being used. In the case of emergency care treatment, (not research in an emergency setting) a wavier will be considered, see section on Waiver of Informed Consent, Alteration of Informed Consent, or Waiver of Documentation for more information.
Children in State Custody (Wards of State)
According to the Department of Children and Families (DCF) applicable policies and by virtue of the court order granting DCF legal custody of certain children (e.g., foster children), that Department is the agency that is authorized to grant permission for participation in research for children in their custody. The decision of whether to grant permission for research is made on a case-by-case basis by DCF and consent is provided by an appropriate representative of DCF.
If a child has begun research procedures with the consent of a parent but is subsequently placed in the custody of DCF while undergoing research interventions, consent must be sought again from the appointed advocate for the child at DCF in order to continue participation in the research. See additional information in Section on Children.
Emancipated Minor
According to Vermont Statute, an emancipated minor means a minor who:
a. has entered into a valid marriage, whether or not such marriage was terminated by dissolution;
b. is on active duty with any of the armed forces of the United States of America; or
c. has been, by a court of law, ordered emancipated.
In order to become an “emancipated minor” the minor must petition the probate court. There are multiple stipulations that must be met by the minor and the court must find that emancipation would be in the best interest of the minor. If the stipulations are met, the court will issue an order of emancipation.
In certain limited circumstances, it may be appropriate to allow an emancipated minor to consent to participate in a research study in the absence of the permission of a parent or legal guardian if the minor has the sufficient capacity to consent to the procedures involved in the research study. Each situation is judged on a case-by-case basis.
9.1.1 Children Reaching Legal Age of Consent While Enrolled in a Study Policy
When a child who was enrolled in research with parental or guardian permission subsequently reaches the legal age of consent to the procedures involved in ongoing research, the subject’s participation in the research is no longer regulated by the requirements of 45 CFR part 46.408 regarding parental or guardian permission and subject assent.
The researcher should seek and obtain the legally effective informed consent, as described in 45 CFR 46.116, for the now-adult subject for any ongoing interactions or interventions with the subjects. This is because the prior parental permission and child assent are not equivalent to legally effective informed consent for the now-adult subject.
As long as the participation continues to meet the regulatory definition of “human subjects research” (for example, it involves the continued analysis of specimens or data for which the subject’s identity is readily identifiable to the researcher), then it would be necessary for the researcher to seek and obtain legally effective informed consent of the now-adult subjects.
A waiver of consent will be considered in those cases in which the subject's continuing participation constitutes no more than minimal risk and meets the other requirements for waiver under 45 CFR 46.116(d), including the requirement that the "research could not practicably be carried out without the waiver." Generally speaking, the UVM IRB will deem it impracticable to re-consent an adult who was enrolled as a child if there are no additional research interventions or interactions. For example:
- Subjects whose parents gave informed consent for their samples to be entered into a biorepository where those samples remain identifiable (either directly or by linking codes).
- Subjects whose parents gave informed consent for their information to be entered into a database where that information remains identifiable (either directly or by linking codes).
- Subjects enrolled as children in interventional trials with parental consent in which all intervention or interaction prescribed by the protocol, including follow-up visits, has concluded prior to subjects' reaching adulthood.
- All subjects' information has been de-identified pursuant to HIPAA such that their data or any samples collected cannot be linked to their identities.
The IRB developed a sample consent/HIPAA template for continued participation in a research study which can be found under the “Consent and HIPAA Guidance” section and should be used when consenting the now-adult subject. This consent form is essentially a continuation cover consent that explains why the now-adult subject is being consented at this time. A copy of the originally signed parental permission and assent (if applicable) should be attached to this continuation consent form and presented to the now-adult subject.. Subjects should be reminded of their right to withdraw from the study including: (a) their right to revoke HIPAA authorization, to the extent that such authorization is revocable under the terms of the informed consent and the authorization signed by the parent(s) or guardian; and (b) their right to revoke any other right granted in the study, (e.g., rights with respect to use of tissue samples) to the extent it would be revocable by the parent(s) or guardian when the subject was a minor.
9.2 Surrogate Consent for Research (Legally Authorized Representatives)
A legally authorized representative (LAR) is defined in both HHS and FDA regulations as an individual or judicial or other body authorized under applicable law to consent on behalf of a potential participant to participate in the procedure(s) involved in the research (45 CFR 46.102(c) and 21 CFR 50.3(1) respectively).
This IRB guidance applies to both therapeutic (with potential direct benefit to the subject) and non-therapeutic (no direct benefit).
IRB REVIEW OF LEGALLY AUTHORIZED REPRESENTATIVES FOR PROVISION OF CONSENT FOR RESEARCH
The IRB evaluates both the consent process and the procedures for documenting informed consent to ensure that adequate informed consent is obtained from participants, unless a waiver of consent has been approved by the Committee. The use of surrogate consent for children or adults with impaired capacity to consent requires additional consideration to ensure effective, voluntary consent to participate.
CHILDREN
See Section 9.1 Children for guidance regarding regulatory requirements specific to consenting children in research.
ADULT SUBJECTS LACKING CAPACITY TO CONSENT
There are no specific federal regulations concerning the inclusion of adult subjects lacking the capacity to consent. These vulnerable subjects have the same rights as other individuals to participate in research, but special care must be taken to ensure adequate informed consent.
Some research protocols involving the cognitively impaired specifically focus on the individual’s condition. Examples include, but are not limited to, patients with dementia, schizophrenia, delirium, intellectual disability, bipolar disorder and stroke. Other protocols may include a variety of subjects and only incidentally include individuals with questionable capacity to consent. In either case, special considerations should be made to ensure that the informed consent process is adequate and appropriate.
The IRB uses a case-by-case approach to approving studies involving surrogate consent. The IRB assesses relevant factors of the proposed study including a potential subject’s consent capacity, the study’s risks, anticipated direct and indirect/scientific benefits, the complexity of the protocol, and the provision of additional safeguards. This type of review process, which is recommended by NIH and based on the framework set forth in Subpart A of the HHS regulations and in FDA regulations on IRBs, is generally a more flexible and comprehensive type of analysis than processes that rely on predetermined categorical criteria, e.g., only studies that provide a direct benefit, or are minimal risk may use a surrogate consent.
ASSESSING CAPACITY TO CONSENT
The researcher must provide details in the protocol submission to allow the determination of whether surrogate consent is allowable. This should include whether the inclusion of subjects with limited capacity to consent is scientifically justifiable (i.e. is the primary goal of the study related to a condition that affects consent capacity; would excluding those with limited consent capacity significantly impact the generalizability of the findings, etc.). The protocol must specify the sequence of steps, and the qualifications of the study personnel, that will be employed to assess capacity to consent and to acquire and document surrogate consent if appropriate.
The assessment of capacity to consent should be specific to the research study. Capacity is generally understood to include at least the following four elements:
- Understanding, i.e., the ability to comprehend the disclosed information about the nature and purpose of the study, the procedures involved, as well as the risks and benefits of participating versus not participating;
- Appreciation, i.e., the ability to appreciate the significance of the disclosed information and the potential risks and benefits for one’s own situation and condition;
- Reasoning, i.e., the ability to engage in a reasoning process about the risks and benefits of participating versus alternatives, and;
- The ability to express a choice about whether or not to participate.
The following methods can be used to assess capacity to consent:
- A standardized and validated instrument that can be tailored to the specific study protocol, such as the MacArthur Competence Assessment Tool – Clinical Research (MacCAT-CR) developed by Appelbaum and Grisso (1995) is available for purchase.
- A post-consent quiz documenting the subjects’ knowledge of critical elements in the informed consent form - i.e., nature of the illness being studied, voluntary nature of participation, ability to withdraw at any time, consequences of withdrawing, possible risks and benefits of participation, procedures involved, time required, confidentiality, and whom to call with any questions. For subjects who score less than perfect on the initial presentation, educational procedures may be employed to raise their understanding to sufficient levels for them to make a meaningful choice about participating. Such procedures may include simple repetition of the relevant information in the consent form or more detailed explanations of items that the subject has difficulty understanding. For examples of educational procedures and the content of such quizzes, see Carpenter et al. (Arch Gen Psychiatry, 2000, 57:533-538), Dunn and Jeste (Neuropsychopharmacology, 2001, 24:595-607), Dunn et al. (Am J Psychiatry; 2001; 18:1911-1913), Dunn et al. (Am J Geriatric Psychiatry, 2002 Mar-Apr;10(2):207-11.), or Wirshing et al. (Am J Psychiatry; 1998; 155: 1508-1511).
- The study investigators may develop and suggest alternative procedures for evaluating the presence of decision-making capacity, - e.g., someone outside the research team making the evaluation as to the potential participant's decisional capacity.
PLAN FOR OBTAINING INFORMED CONSENT FROM SURROGATE
Protocols must include details about the proposed use of surrogates to provide informed consent. Suggested language is below:
- Whenever possible, investigators will attempt to obtain informed consent directly from the subject.
- If the potential research subject is unconscious or otherwise obviously lacking in decision-making capacity, the investigator shall document that observation in the research record.
- The researcher will document the process used to determine who was able to provide surrogate consent.
- If the potential research subject has questionable capacity to consent but is not unresponsive, the investigator will describe the research to the subject and perform and document an assessment of the participant’s capacity to provide consent.
- If lack of decisional capacity is evident, the investigator shall inform the potential research subject of the investigator’s intent to obtain surrogate consent. If the subject expresses resistance or dissent to either study participation or to the use of surrogate consent they will be excluded from the study.
UVM MEDICAL CENTER POLICY ON SURROGATE CONSENT
The IRBs for UVM and The UVM Medical Center use the same standards for determining who can provide surrogate consent for research.
Specifically, consent may be given by:
- The decision-maker designated by the patient in a Living Will or Durable Power of Attorney for Health Care (“Advance Directive”);
- A judicially-appointed guardian if the court order explicitly grants the guardian clinical decision-making authority pursuant to 14 V.S.A. § 3069 (c ) (2);
- If the patient has not designated a decision-maker in his/her Advance Directive, consent may be obtained from a spouse, civil union partner, adult child, adult sibling, or other family member/friends. Providers should exercise their professional judgement when determining which family member(s) should be consulted as the patient’s surrogate decision-maker. In general, when determining who should serve as the surrogate decision-maker, providers should assess which individuals best knows what the patient would want in a given circumstance (i.e. the individual best equipped to offer a substituted judgement)
Please note that medication to treat psychiatric illness is not an allowable intervention when a potential subject is unable to give informed consent.
Additional Considerations
Per FDA regulations: A verbal approval does not satisfy the 21 CFR 56.109(c) requirement for a signed consent document, as outlined in 21 CFR 50.27(a). However, it is acceptable to send the informed consent document to the legally authorized representative (LAR) by facsimile and conduct the consent interview by telephone when the LAR can read the consent as it is discussed. If the LAR agrees, he/she can sign the consent and return the signed document to the clinical investigator by facsimile.
If a study that involves subjects who lack the capacity to consent takes place over extended periods of time, the researcher should consider whether and when periodic re-consenting of individuals is required to assure that a subject’s continued involvement is voluntary. The IRB may require that the Investigator re-consent subjects after taking into account the study’s anticipated length and the condition of the individuals to be included (e.g., subjects with progressive neurological disorders). Additionally, the IRB considers whether and when to require a reassessment of decision-making capacity.
9.3 Cases of Physical Compromise
Revised 10/23/2023
A person who can understand and comprehend English, but is physically unable to talk or write, can be entered into a study if they are competent and able to indicate approval or disapproval by other means. If (1) the person retains the ability to understand the concepts of the study and evaluate the risk and benefit of being in the study when it is explained verbally (still competent) and (2) is able to indicate approval or disapproval to study entry, they may be entered into the study. The consent process should document the method used for communication with the prospective participant and the specific means by which the prospective participant communicated agreement to participate in the study. An impartial witness must be present during the entire consent process and sign the consent document. Refer to the definition of impartial witness in section 29.
This consent process must be approved prior to use. The protocol submission must:
1. Detail how the participant will be assessed for their comprehension and understanding of the research.
2. Explain why the participant is unable to sign their name and date the consent form.
3. Ensure an impartial witness will be made available to witness the discussion and the agreement of the participant to enroll in the protocol.
4. The impartial witness will sign the consent.
Below is an example of the signature page that may be appended to the proposed consent in these situations.
_________________________________________ _________
Signature of Participant (mark here with "X" if unable to write) Date
_________________________________________
Name of Participant Printed (research staff may complete if participant is unable to write)
_________________________________________ _________
Impartial Witness (to be used in the event the participant is unable to write) Date
_________________________________________ _________
Signature of Principal Investigator or Designee Date
_________________________________________
Name of Principal Investigator or Designee Printed
9.4 Informed Consent and HIPAA Authorization Process for Non-English Speaking Individuals
Revised 12/07/20
The Department of Health and Human Services (DHHS) regulations for the protection of human participants require that informed consent information be presented in "a language understandable to the participant" and in most situations, that informed consent be documented in writing (45 CFR §46.116 and §46.117).
For non-English speaking participants to participate in a research study, steps must be taken to assure true informed consent is obtained. This does not simply mean a form is signed, but rather steps are taken to assure study procedures and risks are understood by the participant. An interpreter may need to be involved in the informed consent discussion as well as a translated consent document. Further, the IRB may require the investigator to submit a back-translation of the informed consent.
Definitions:
- Back translation: process of translating written materials from one language to another and then, in a separate process, translates the document back into the original language. This process is performed to evaluate the quality and integrity of the information being translated.
- Certified Translator: a professional translator who has successfully completed a certification program or exam providing them with certified translator credentials
- Interpreter: person who accompanies researchers, in real time, to convey verbal information to another person in their native language
- Medical Interpreter: an interpreter who is familiar with medical terminology
- Non-English speaking: unable to comprehend English language
- Translator: person who converts written materials from English to another language
- Witness: an individual who is fluent in both English and the necessary foreign language who will be physically present during the consent process to observe the process and sign consent forms
Long Form Consent and HIPAA Authorization process §46.117(B)(1)
When the participant population of any research study is expected to include a significant number of participants who are not fluent in English but are fluent in another language, the IRB requires a full translation of the English version of the approved consent document (Long Form) along with the translator’s documentation of qualifications.
IRB Review for the Long Form process
In addition to the standard initial submission review materials, submit the following:
1. English language version of consent. Documents should first be submitted to the IRB in English, and once approved, be sent to the translator. A modification should then be submitted to provide the translated documents.
2. A plan for ensuring the participant understands the requirements and the voluntary nature of the research
The following items must be resubmitted for final approval of the translated documents:
1. Participants primary language version of the consent.
2. Documentation describing the qualifications of the translator and the date of translation.
Long Form Consent Documents – Request for Back-Translation
If the research is deemed “high” risk or is very complex, or there are other relevant concerns, the IRB reserves the right to request the translated consent be translated back into English to ensure the translated consent is written in understandable terms and contains all key elements of consent.
The IRB requires documentation that this back-translation was done by a different translator than the one who did the original translation and documentation of that second translator’s qualifications is required as well.
Consent Process, Signatures and Record-keeping Requirements with Translated Long Form
1. Conduct the participant's informed consent process with the researcher and an interpreter fluent in both English and the participant’s primary language.
2. The participant signs the translated informed consent.
3. The researcher or designee signs the translated informed consent (interpreter may interact in person, by phone or video-conferencing and does not need to sign the consent form).
4. The participant is given a copy of the translated informed consent.
5. A copy of the signed documents is maintained in the study records.
6. A copy of the signed documents should be included in the participant's medical records if that is the standard practice for this study.
Oral Translation with Short Form Consent and HIPAA Authorization process
This method is used for the occasional and unanticipated non-English-speaking subject meets enrollment criteria and wishes to participate. Given the unexpected nature of this situation and the steps that must be taken to prepare for this type of alternative consent process, it is not always possible to obtain consent on the same day a potential non-English speaking participant is identified.
§46.117(b)(2) Short form consents are generic research consent forms that can be translated into multiple common languages and are limited to the basic elements of consent. The UVM IRB has provided a diverse set of short form consents in a variety of languages translated on our website for researcher convenience.
A short form written informed consent stating the elements of informed consent as required by §46.116 must be presented orally to the participant or the participant's legally authorized representative, and the key information required by §46.116(a)(5)(i) is required to be presented first to the participant, before other information. The IRB shall approve a written summary of what is to be said to the participant or the legally authorized representative. When this method is used, there shall be a witness to the oral presentation. Only the short form itself is to be signed by the participant or the participant's legally authorized representative. However, the witness shall sign both the short form and a copy of the summary, and the person obtaining consent shall sign a copy of the summary. A copy of the summary shall be given to the participant or the participant's legally authorized representative, in addition to a copy of the short form.
Typically, this option is used when a single participant is found to be eligible to participate in research and there is no long form consent translation. A witness to the oral presentation is required.
The Short Form process should also be used when enrolling a non-English speaking participant who may not have a written language (ie. Mai-Mai) that can be translated into a short form consent. In this case, an interpreter will read the oral summary of consent procedures, risks, objectives to the participant but there will be no translated short form to sign. All parties taking part in the consent process will sign the English version consent form. It is imperative that the research team has good consent process documentation to ensure legally effective consent in this rare case.
IRB Review for the Short Form process
Submit the following for review with the modification request in UVMClick-IRB:
1. English version of short form consent (template located on our forms page).
2. Translated version of short form consent.
3. Documentation describing the qualifications and date of translation (if not using an already approved translated language provided on UVM’s IRB website.)
4. Prepare a written summary of the study-specific details that will be orally communicated to the subject by the Interpreter.
Note: The IRB-approved English language consent form may serve as this summary and the IRB encourages the use of the approved consent to serve this function.
5. If PHI is to be obtained as part of the research then a request for an alteration of HIPAA will need to be completed in the UVMClick record as well.
Expedited review of these versions is acceptable if the protocol, the full English language informed consent document, and the English version of the short form document have already been approved by the IRB.
Consent Process, Signatures and Record Keeping Requirements When Using a Short form Consent Document
Per the Office of Human Research Protections (OHRP), the following steps must be followed after IRB approval.
- The participant reads the translated short form consent document in their native language
- Interpreter presents the oral version of the IRB-approved English consent form (or written summary of study-specific details if the Investigator has decided not to use the IRB-approved English consent form to meet the oral presentation requirement)
- A study team member, who is approved to obtain consent, must be present for this presentation.
- If the Interpreter is not also acting as the Witness, the Witness must be present during this presentation as well
- The Interpreter facilitates participants asking questions and study team members providing answers, to ensure participant understanding
- When all of the Participant's questions and concerns have been addressed, the Participant signs and dates the translated "Short Form" consent document
- The researcher signs the IRB-approved English version of the informed consent document
- The witness (fluent in both languages) signs BOTH the translated short form and the written English consent version. (Note, when the person obtaining consent is assisted by an interpreter, the interpreter may serve as the witness.)
- The Participant receives copies of both consent forms. If the study is FDA regulated, the participant must receive signed copies of both consent forms.
- A copy of the signed documents is maintained in the study records along with clear documentation of the consent process and who was involved. (consent process documentation form is available on the IRB website)
- A copy of the signed documents should be included in the participant's medical records if that is the standard practice for this study.
Any deviation from these alternatives requires review and approval by the IRB.
Effective Communication During Study Participation
For consent to be legally effective, the participant must be provided with all relevant research-related information and must clearly understand such information. When non-English speaking participants are being invited to participate in a study, investigators must ensure that there is adequate communication between the research team and the participants.
Unless the principal investigator or a member of the research team is fluent in the prospective non-English speaking participant’s language, an interpreter will be necessary to facilitate the conversation during the consent process and for communication throughout the course of the study. Interpreters should be fluent in English as well as in the language of the non-English speaking participant.
Other Considerations
Recruitment materials such as flyers must be translated in order to accommodate expected non-English speaking participants (i.e., a significant number of participants who are not fluent in English). All translations of recruitment materials must be completed by a certified translator and approved by the IRB prior to their use.
Study instruments may be in English and translated orally by an interpreter or a member of the research team who is fluent in the language spoken by the non-English speaking participant. If an investigator prefers to have any study instruments translated, the translations must be completed by a certified translator and approved by the IRB
Interpreting & Translation Services
Language Access Services at the University of Vermont Medical Center offers interpreting and translation services for patients with hearing loss and patients with limited English proficiency. These services should be used when interacting with participants involved in clinical research. Researchers can access on-site interpreters in many languages. They have telephone and video remote interpreters available 24 hours a day. Researchers can also request translation of research documents. Language Access Services can be reached at UVMMC Language Access Services.
Note: Interpreter & Translation Services at UVMMC meet the standards, therefore, additional documentation of qualifications is not required. We do, however, require documentation of qualifications for any other translator services.
The cost of translating written consents is the investigator's responsibility. These costs may be quite high, particularly for large studies where multiple translations are needed and/or studies with relatively complex consent information that may require additional time by a skilled professional. Investigators should include the costs of written translations as well as medical interpreter services on grants and contracts as applicable. Industry sponsors are often willing to pay the costs of translating consent forms. The UVM IRB does not endorse any specific interpreter & translation service. The following are additional services researchers have used previously.
9.5 Waiver of Informed Consent, Alteration of Informed Consent, or Waiver of Documentation §____.116(c)
Non-FDA Regulated Research
Waivers: 45 CFR 46.116(f)(1) In some research, written or verbal consent is not possible. A typical example would be retrospective record or pathology reviews for limited information that is not sensitive in nature and the data are derived from clinically indicated procedures. Consent is not possible because the subjects are not available to sign a consent form.
Alterations: 45 CFR 46.116(f)(2) In some research, an alteration of the individual’s informed consent or elements of the process may be waived. An example would be when research requires deception. In these cases, some of the elements of informed consent are met but not all. Information typically held would be the basis for the research and subjects are later debriefed.
45 CFR 46.116(f)(3) The requirement to obtain written or verbal informed consent may be waived or altered if the IRB can find and document that:
i. The research involves no more than minimal risk to the subjects;
ii. The research could not practicably be carried out without the requested waiver or alteration;
iii. If the research involves using identifiable private information or identifiable specimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format;
iv. The waiver or alteration will not adversely affect the rights and welfare of the subjects;
v. Whenever appropriate, the subjects or the legally authorized representatives will be provided with additional pertinent information after participation.
In the case of Emergency Use for Treatment, a waiver will be considered, see applicable section below.
The IRB will evaluate the request to ensure that the waiver criteria set forth above are met. If the waiver is granted, then the waiver approval signed by the IRB chair or designee shall be returned to the principal investigator.
Note: If the protocol requires protected health information (PHI) and you are applying for a waiver of consent, then a waiver of authorization must also be requested. Consent/Authorization/Documentation. If there is no protected health information, HIPAA regulations do not apply and a waiver of authorization would not be required. See additional information under the Health Information Portability and Accountability Act section.
FDA-Regulated Research
FDA regulations have historically not permitted any waiver or alteration of consent only under very strict conditions as described below and in these two regulatory sections Emergency Use of a Test Article 21 CFR 50.23(link is external) and Emergency Research 21 CFR 50.24(link is external).
In July 2017, FDA released new guidance IRB Waiver or Alteration of Informed Consent for Clinical Investigations Involving No More Than Minimal Risk to Human Subjects(link is external) to allow for waiver of informed consent only for certain FDA-regulated minimal risk clinical investigations. This is to facilitate investigator’s ability to conduct studies that may contribute substantially to the development of products to diagnose or treat diseases or conditions, or address unmet medical needs. The FDA intends to revise its informed consent regulations to add this waiver or alteration under appropriate human subject protection safeguards to the existing exceptions from informed consent. However, until FDA promulgates these regulations, the FDA will not object to an IRB approving a waiver or alteration of consent under the criteria noted previously.
Emergency Use for Treatment (21 CFR 50.23)
If your protocol includes an FDA-regulated test article (drug or device), an exemption from the consent requirement is permitted for “Emergency Use for Treatment”. See section Emergency Use of an Investigational Drug or Biologic or Investigation Device for more information.
Emergency Research (21 CFR 50.24)
Emergency research is planned research involving human subjects who have a life-threatening medical condition that necessitates urgent intervention (for which available treatments are unproven or unsatisfactory), and who, because of their condition (such as traumatic brain injury) cannot provide informed consent.
Proposed research activities which contain an exception to consent for emergency research, referred to as an “Emergency Research Consent Waiver,” must meet the strict limited conditions set forth by FDA regulation.
Waiver of Documentation of Informed Consent
45 CFR 46.117(c)(1) In some research, verbal or implied consent of the subject is sufficient and a signed consent form is not necessary. A typical example would be a mailed survey with a cover letter explaining the research. The receipt of a completed survey implies that the subject wanted to participate. The requirement for the investigator to obtain a signed consent document from some or all subjects may be waived if:
i. The only record linking the subject and the research would be the consent document and the principal risk would be potential harm resulting from a breach of confidentiality. All subjects, however, must be asked whether they want documentation linking them with the research. The subject's wishes will govern and should be adequately documented, regardless of final decision.
OR
ii. The research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside the research context
OR
iii. If the subjects or legally authorized representatives are members of a distinct cultural group or community in which signing forms is not the norm, that the research presents no more than minimal risk of harm to subjects and provided there is an appropriate alternative mechanism for documenting that informed consent was obtained.
(2)In cases where the written documentation requirement is waived, the investigator may wish to provide the subjects with a simply written statement or information sheet that describes the research. This written statement must be reviewed and approved by the Committee prior to use. See forms page on our website for template. For protocols where verbal consent is obtained, the principal investigator or a research team member must document in the research file that verbal consent was obtained. See forms page on our website for consent process documentation form.
UVM IRB Approved Waivers Cannot be Used at Other Institutions
Waivers approved by UVM are specific to entities under the UVM IRB purview, which are UVMMC and the UVM Luse Center. UVM waivers are not recognized by other covered entities and therefore, cannot be applied to other hospitals or clinics. Subjects would need to complete a medical release form from that hospital or clinic or you need apply for a waiver from an associated IRB at that institution.
9.6 Consent Process for Legally Blind or Impaired Vision Research Participants
For subjects who cannot read the consent materials due to blindness, or the subject's legally authorized representative (LAR) is legally blind, the following the consent process is recommended:
- The consent and HIPAA authorization forms should be presented to potential subjects orally and the potential subject provided with an audiotape or videotape of the process if possible.
- Have an impartial witness observe the consent process, such as a subject advocate or someone not affiliated with the research team. The witness must be a person who is independent of the research team and cannot be unfairly influenced by people involved with the research, who does not have a coercive relationship with the participant, who attends the informed consent process.
- If potential subjects have access to equipment that can read the consent document for them, provide sufficient time for them to review the consent document independently of the research team.
- As would be expected for any consent process, ensure sufficient time is allowed for questions to be asked by the potential subjects, subject's representative, and research team to ensure that the consent process was clear and effective.
- The research documents should include a statement that the forms were read to the subject by a member of the research team designated to obtain informed consent.
- It is the investigator's responsibility to judge the subject's comprehension of the consent information including the understanding the participation is voluntary and that that subject has the right to withdraw at any time during the study. If the investigator doubts the subject's consent comprehension, he or she should not enroll the subject in the study.
Example signature block for witness signature
Add on as needed to the last page of the consent if a witness will observe the consent process. E.g., blind subject. Do not add to every consent document unless every subject will have a witness to the consent process.
| Signature Block |
|---|
My signature below documents that the information in the consent form and any other written information was accurately explained to, and apparently understood by, the subject, and that consent was freely given by the subject. |
Signature of witness to consent process |
Date |
9.7 Guidance for Researchers Using Deception or Incomplete Disclosure in Research
Revised: 4/5/22
This guidance describes the special responsibilities imposed on the investigator and the considerations required of the IRB when research involves deception or incomplete disclosure.
Deception studies intentionally provide misleading or false information. Misleading or omitted information might include the true title of the research, the purpose of the research, the role of the researcher, performance feedback given during the research, or what procedures in the study are actually experimental. Deception and concealment increase ethical concerns because they interfere with the ability of the participant to be fully informed at the time of consent. However, deception may be necessary in some cases.
Examples of deception include:
• Participants complete a quiz, and are falsely told that they did poorly, regardless of their performance.
• An anxiety study, in which participants are told to expect mild pain during the course of the study, but no painful procedures are administered.
Examples of Incomplete disclosure include:
• Participants are asked to take a quiz but they are not told the research question involves how background noise affects their ability to concentrate.
• Participants are told they are completing a survey to evaluate customer service when the true purpose of the study is to correlate psychological responses with patient care satisfaction.
It is important to note that in some cases the IRB does not consider withholding the true hypothesis from the participant as significant deception.
Regulations
Federal regulations permit but establish limitations on the use of deception. The Investigator must provide for IRB review and approval their scientific and ethical justification for deceptive procedures. The missing information should not increase the risks to participants in the study, and when possible, participants must be fully debriefed as soon as the research methodology allows. Participants must have the opportunity to ask questions about the new information and must be given the opportunity to withdraw from the study as well as the opportunity to have their data removed. Deception may not be utilized to obtain enrollments.
According to guidance provided by the Office for Human Research Protections (OHRP), the use of deception requires a request to waive certain elements of informed consent. According to the regulations the IRB may approve a consent procedure, which does not include, or which alters, some or all of the elements of informed consent IF it finds and documents that:
(1) The research involves no more than minimal risk to the subjects;
(2) The waiver or alteration will not adversely affect the rights and welfare of the subjects;
(3) The research could not practicably be carried out without the waiver or alteration; AND
(4) Whenever appropriate, the subjects will be provided with additional pertinent information after participation.
(5) The research is not FDA-regulated.
To apply for an alteration of the consent process to waive certain elements of consent, the Principal Investigator must check yes to question #2 on the UVM Consent/HIPAA Information page in UVMClick and attach the debriefing script (if appropriate). The IRB will evaluate the request to ensure the alteration criteria set forth above are met. If the alteration of consent processes is granted, this will be reflected in the approval notification.
Consent Form Considerations
If the study design allows, participants should be told during the original consent process that some information is being withheld or is incomplete, and that they will receive more information after the research is over. However, researchers often believe that even vague references to hidden purposes will affect the participants' behavior and make the study impracticable. Investigators should either add such language to their consent forms when it is possible or note in their protocols why it is not feasible to do so. The consent form can omit information, but investigators should avoid deliberate misinformation or falsehoods within the consent.
Exempt Research
Deception is allowable under Benign Behavioral Interventions 45 CFR 46.104(d)(3)(iii).
If the research involves deceiving the subjects regarding the nature or purposes of the research, this exemption is allowed only if the subject authorizes the deception through a prospective agreement to participate in research in circumstances in which the subject is informed that he or she will be unaware of or misled regarding the nature or purposes of the research.
9.7.1 Debriefing
When an investigator uses deception or incomplete disclosure, regulations require that participants be debriefed as soon as possible after participation in the study, or at the end of the study with appropriate justification provided. Requesting to delay debriefing to the end of the study and may require a Full Committee Review. Participants should be given a simple, clear and informative explanation of the rationale for the design of the study and the methods used, and participants should have the opportunity to ask questions. There are rare cases when debriefing may be impossible or may do more harm to research participants than the deception itself – in those cases the IRB may decide that debriefing is not appropriate. Click here for ![]() UVM’s debriefing template (DOC).
UVM’s debriefing template (DOC).
The primary goals of a debriefing process are to:
- inform participants of the true goals of the research study,
- remove any effects of false information they were given,
- educate participants about the research process, why deception is sometimes necessary
- make participants feel that they are an important part of the research process.
The investigator may decide (or the IRB may require) that the debriefing include an option for participants to withdraw their data from the study after they learn the true nature of the research, if it is of a particularly sensitive nature. (e.g., the withheld aim of the study is that the researcher is measuring participants’ racism).
9.7.2 Additional IRB Considerations when using Deception or Incomplete Disclosure in Research
The American Psychological Association (APA) has also developed a code of conduct for deception in research. According to the APA, deception in research requires that the researcher:
(1) apply a cost-benefit analysis that explicitly considers the potential for harm created and/or exacerbated by the use of deception,
(2) consider alternative methods, and
(3) fully explain the nature of the deception at the conclusion of the study or explicitly justify withholding such information. In all cases, the safety and comfort of the participant should be of paramount concern.
When IRB members are evaluating the use of deceptive techniques, IRB members should be considerate of the APA’s code of conduct as well as the following:
- The scientific value and validity of the research
- The efficacy of alternative procedures
- The certainty that deception does not extend to influence participants' willingness to participate
- The possibility of experimentally induced harm and the ability of the proposed procedures to remove such harm through debriefing
- The potential of deception to facilitate unwanted and inappropriate invasions
9.8 Media Consent
If a subject consents to participate in a protocol and then agrees to be interviewed regarding their research participation, another consent document must be signed. This consent document is referred to as “media” consent. It is required because the originally signed protocol consent form states that the subject’s information will be protected and kept confidential. The “media” consent indicates that they are freely giving up that protection by agreeing to take part in the interview. This applies when there is a direct or indirect interview, videotaping, and photographs of the individual for TV/radio broadcast or publication. This “media” consent is located on our website forms page and should be completed by the subject prior to the interview. The IRB does not need to review this activity.
9.9 Obtaining Electronic Written Consent
Version 12/09/20
Electronic consent (eConsent) is the use of electronic systems and processes, whether in-person or remotely, that employ multiple electronic media (e.g., text, graphics, audio, video, podcasts, websites, etc.) to convey information relating to a research study and to document informed consent of participants who wish to participate in a study. eConsent has been sanctioned by both the Department of Health and Human Services (HHS), Office of Human Research Protections (OHRP), and the Food and Drug Administration (FDA).
The purpose of this guidance is to describe eConsent requirements for research studies at UVM/UVMMC. Although compliance with applicable laws and regulations is necessary, such requirements will not be arbitrarily more stringent than those for obtaining informed consent in a traditional face-to-face interaction with documentation by signature on a paper consent document. Participant populations involving surrogate consent, non-English speaking individuals, children or those with physical disabilities may be consented electronically. Specific consent requirements that apply to populations considered vulnerable still apply and can be found in the IRB Policy and Procedures document.
eConsent may be used in the following ways:
- Full and expedited studies may use eConsent in person to either supplement or replace paper-based informed consent processes that address participant’s needs and/or preferences. Requests to use eConsent remotely for full studies will be reviewed on a case-by-case basis.
- Expedited studies may use remote eConsent following IRB-approved remote consent processes (e.g. video/telephone.)
- Exempt projects may use remote eConsent by providing links to post QR codes, web-links on study posters, brochures, or websites; email link.
The IRB is responsible for ensuring that the proposed eConsent process is appropriate for the level of risk and the population engaged in the research. The IRB may decide that informed consent must be obtained face-to-face.
IRB eConsent Submission Requirements
Process Details
Provide the following details in the protocol regarding the proposed eConsent process.
- How will eConsent be provided, signed and collected;
- Explain how the eConsent will be provided to participants. If emailing or texting, the study team must obtain verbal permission to send the eConsent via email or text. Example: “Because UVM can’t control the security of email or text messages once we send them, we need your permission to text or email you. Do you want to receive the link to the eConsent via text or email?” The permission should be documented. The email/text should not include PHI. Provide proposed introductory email/text language.
- Identify how the subject signatures will be obtained (typed signature, PIN number, written signature – via stylus/cursor, etc.);
- How will you verify the person who is enrolling is the person consenting;
- If any or all of the consent process takes place remotely and is not personally witnessed by study personnel, the process must include a method for identity verification to ensure that the person electronically signing the informed consent is the participant who will be participating in the research study. See Technical Guidance for different options to authenticate participants.
- Will consent be self-guided or led by a study team member;
- Provide details of the support being provided to the participants.
- Systems that support eConsent must be easy to navigate, allowing the user to proceed forward or backward, the option to stop and continue later. Systems should incorporate electronic strategies to encourage a participant to access all the consent material.
- How will participants get questions answered before consent;
- This may be accomplished by scheduling in-person discussions with study personnel or through a combination of electronic messaging, telephone calls, video conferencing or electronic chatting.
- Will an assessment of consent understanding be included; and
- How will the participant receive a copy of the signed consent?
- HHS, FDA, and the HIPAA Privacy Rule, as applicable, require that the subject/subject’s legally authorized representative be provided a copy of the written informed consent, unless the documentation of written informed consent has been waived.
- While the FDA regulations do not require that the subject’s copy include a signature, FDA recommends that a copy of the signed informed consent form that includes the date when the eConsent was signed be provided to the subject.
- If PHI is used the project, the HIPAA regulations require that the subject/subject’s legally authorized representative receive a signed copy of the consent form because the authorization is included within the consent form.
- The copy provided to the participant can be paper or electronic and may be provided on an electronic storage device or via email. If the copy provided includes one or more hyperlinks to information on the Internet, the hyperlinks should be maintained, and information should be accessible until study completion (if a paper version is provided, it should contain the necessary content from any hyperlinks)
- Participants must be informed in the consent form that any correspondence via email are not considered secure.
- When the study team will not physically interact with the subject, the software should be set up to display a button for the subject to download the signed consent form. If this is not possible, other methods need to be used to provide the subject a signed copy (e.g. paper copy through the mail, emailed PDF).
HHS and FDA have developed joint guidance on electronic consent which is found here https://www.fda.gov/regulatory-information/search-fda-guidancEdocuments/usEelectronic-informed-consent-clinical-investigations-questions-and-answers.
Consent Forms or Other Media to Supplement Consent
Researchers must submit to the IRB the proposed consent and any supporting consent media. Whether utilizing a consent form or using of other types of media, all required elements of informed consent must be included, unless the IRB has waived one or more of the elements.
- Create WORD versions of all consent documents and submit your study application to the IRB as usual.
- Keep in mind when developing the WORD consent that any checkboxes for optional participation and questions built into the system to gauge subject comprehension should be inserted within the WORD consent document and noted as such. Any methods for verifying identity of participant can also be built into the consent at the end of the document and noted as such.
- Remember to include language in the consent document notifying participants that they will receive a signed copy, if applicable, via the email address they provided. Email is not a secure means of communication so this should be disclosed as a potential risk for loss of confidentiality and participants should be aware when they provide consent.
- Inclusion of the “person obtaining consent” in the eConsent is required when consenting participants in clinic, however, adapting to eConsent, this may not be necessary.
- Once your protocol and consent documents are approved, the watermarked version of your consent form will be available in Click IRB under your newly approved study.
- If other forms of media will be used, (e.g. videos, web-based presentations) those items must be submitted for IRB review. These materials can be submitted in WORD format or by providing a link.
IRB Approval of the eConsent and any Associated Consent Media
As part of the approval process of the electronic consent you will be required to build the approved WORD consent into either RedCap or Qualtrics (approved institutional software for research) and provide a link for IRB review. The IRB will be assessing the functionality of the eConsent and whether the eConsent version reflects the IRB-provisionally approved version. Approved, optional checkboxes, assessment questions and/or authentication questions will be confirmed as being present as well as areas for signature.
Building an eConsent
Please see section 9.11 Technical Guidance for specifics, additional information and links to how to build eConsent.
The priority is to ensure that the final product/process be easy for the participant to navigate, read and understand. Additionally, it is important that study staff, as well as IRB staff, can readily determine which version of the eConsent is being used at any given time.
There are currently two options to build an eConsent. You may create pictures/images of each page of the IRB approved consent to load in either RedCap or Qualtrics or you may copy and paste IRB approved consent text from the approved WORD document into the software, along with the IRB approval date in the header and version date in the footer. Things to consider when deciding how to build an eConsent.
Images Option
- Since the images include the actual IRB stamped consent form generated from UVMClick, researchers can verify the most recent version is being used when consenting participants. For every modification to the consent, you need to screen print each individual page and upload the full consent form to reflect the new approved dates.
- Different browsers may skew images or may not allow images.
- Consent process documentation is required.
Text Option
- In this option, the IRB stamp is not scanned, so researchers need to ensure the IRB approved date and consent version dates are entered accurately. We believe this may be an area that is prone to compliance issues if not done correctly. Always verify the most recent approved version by checking within the UVMClick record.
- For every update to the consent, the appropriate section or sections need to be replaced and the IRB approval date and consent version date need to be updated. Researchers must also be careful not to alter areas of the consent that have been approved previously. This activity would be prone to compliance issues if not done correctly.
- Text formatting is typically user friendly with regards to adjustments across browsers. This may be preferable when participants will be reading the consent on a small tablet or phone.
- You must provide a link to a PDF of the IRB-stamped version for participants to download.
- Consent process is required and must include the version date of the approved consent form.
Modifications to the eConsent
Projects requiring a change to the eConsent would need to update the last approved WORD consent document and submit through UVMClick as usual. Once your consent modification is approved, you will need to submit a clarification response when you have updated the eConsent to provide a link back to IRB staff for final approval.
Consent Process Documentation
Just as is the practice with in-person consent, the investigator must document the consent process using one of the consent process documentation examples. The documentation template should be modified to capture all the above information (e.g. version date of consent, how contacted, how identity ascertained, date/time, etc.).
In addition to the system maintaining documentation of participant’s signed eConsent, Investigators need to maintain the fully executed consent and the consent process documentation in the participant’s research record. This is Good Clinical practice and may be reviewed as part of a quality assurance visit.
eConsent and Waiver of Documentation of Written Consent
Regulations (under 45 CFR 46.117(c) and 21 CFR 56.109(c)) allow for a waiver of documentation of consent (e.g. no signature of the participant) for those studies where there is not more than minimal risk/risk is commensurate with daily living. This is true for eConsent as well.
Example: Participant receives email with explanation (information sheet) about research study (approved by IRB). The email includes a link to agree to participate and/or contact information if the person has questions. Once consent has been obtained electronically within the system, the participant is brought to the research survey. In this instance, the PI does not speak with the individuals unless they have questions about the research.
The system maintains the documentation of participation.
FDA Inspection Requirements
During inspections of clinical investigation sites, FDA regulations require that FDA be granted access to records and reports made by the investigator, including site-specific versions of the eConsents, the materials submitted to IRBs for review and approval, all amendments to the site-specific eConsents, and all subject-specific signed eConsents. These should be available at the site either in electronic or paper form. FDA reserves the right to review the content of the eConsent program or informed consent document and the corresponding informed consent of the subject/subject’s authorized representative and the signature of a witness, where applicable, along with the date that the eConsent was signed. Any updates to the documentation should also be available for review.
9.10 Telemedicine and Research Visits
Revised 1.11.2023
Since the pandemic there has been an exponential increase in the delivery of care via telemedicine and other digital means. This guidance is being updated with legal, regulatory, technical, and operational considerations to keep in mind when planning research that incorporates telemedicine.
Vermont Law
The IRB must follow the statutory requirements for telehealth used for delivering health care services through telemedicine under Vermont Law [18 V.S.A. 9361]. *Please note that while Vermont law does not preclude telehealth to be used for research it does not allow any telehealth visits to be recorded. This prohibition applies to both clinical healthcare visits as well as research visits that include any clinical aspects. Research required visits conducted virtually that do not involve clinical aspects (e.g., surveys, interviews, focus groups) may be recorded.
IRB Submission Requirements
Proposals to conduct virtual interaction or intervention must include the following items.
Contact Information
The study team must clearly explain how contact information such as emails or telephone numbers will be sought for virtual participation.
Consent
Consent to virtual research participation must be obtained from the participant prior to their participation. The consent may be written or oral with documentation of consent process in the research record. The consent/consent script must include the following points as applicable:
• Informing and obtaining permission of the participant to allow the presence of any other individual who will be participating in or observing the visit;
• Assurance the visit will be delivered over a secure connection that complies with regulations, and for PHI, specifically the requirements of the Health Insurance Portability and Accountability Act.
• Informing the participant that because there is virtual access to the participant’s environment, the researcher may witness things that he/she may be required to report to authorities such as abuse, child endangerment, drug use or other illegal activities.
Protocol Language
The protocol must reflect the procedures that will be used during the visit. This includes how consent will occur, whether the participant will be alone or with another person or group, what will happen during the visit, which software program will be used and whether there is intention to record the visit.
Software/Programs
If data is to be collected virtually, the study team must explain which communication streams (e.g., Zoom through UVMMC, zoom through LCOM or Microsoft Teams) will be used for these purposes. See Technical Guidance for Remote Visits, Electronic Consent, and Data Capture.
Virtual Observations
• If virtual classroom observations are proposed, the study team must extensively detail the platforms used by the schools and how access to these virtual classrooms will be arranged.
• Virtual observations will provide access to the participant’s home environments, background conversations between family members, presence of non-participant children, etc. The study team must clearly explain what measures will be in place to avoid collecting this data.
• If virtual observations require recording procedures (only allowed for nonclinical visits), clear explanation of how only consented participants will be recorded will be required.
Individual Interviews
If individual interviews with children are to be carried out virtually, the study team will be required to account for how the safety of the child (e.g., conducting the interview in the presence of an adult) will be ensured without compromising the privacy of the participant.
Focus Groups
If focus groups are to be conducted virtually, provisions to prevent recording by participants and, where applicable, safeguard child participants, must be accounted for.
Recordings
• A clear explanation of what will be recorded and how the recording will be protected.
• If only audio-recordings are proposed, the study team must attest that video-recordings will not be made.
• Vermont law prohibits telehealth visits to be recorded for clinical or research purposes where clinical aspects are included.
Data Security
A clearly described account for how research data will be stored and safeguarded under work from home conditions will be required.
Newly Identified Risks
• A shift to virtual data collection will necessarily entail additional collection of private identifiable information to ensure the participant is the correct participant (for example you may ask for a driver’s license.) The modification must clearly account for any/all additional risks this will pose and what measures will be in place to mitigate or reduce these risks.
• Access to a study subject’s home may yield reportable or otherwise sensitive, illegal, or damaging information (e.g., seeing drugs or drug paraphernalia in a child’s home, witnesses domestic abuse, gaining access to a home that may otherwise appear unkept or hazardous, etc.) The study team must clearly account for these additional risks in all consent documents, and further explain how they will be reduced or mitigated.
• Virtual meetings carry risks such as Zoom bombing or related, which must be accounted for and reduced or mitigated.
9.11 Technical Guidance for Virtual Research Visits, Electronic Consent, Electronic Data Capture, and Communications
New 12/09/20
Approved Software and Appropriate Use
UVM’s Compliance Office and UVM/LCOM technical services are required to review and approve software systems that support research efforts. The following is a list of currently approved software and their appropriate uses for both UVM and UVMMC Health Network.
Virtual Research Visit Software
- UVMMC HN – The hospital Zoom license is HIPAA compliant. Researchers must use their uvmhealth.org ID to utilize this program.
- UVM LCOM – The Larner College of Medicine Zoom license is not HIPAA compliant (does not have a Business Associate Agreement in place); however, the program has settings to mirror the minimum criteria for compliance when PHI is involved. Use that includes PHI is allowed if these settings are strictly adhered to. Settings to allow LCOM Zoom license use are located below.
- UVM – Teams may be used when there is no PHI involved.
Determining Which Program to Use for Remote Visits
Determining which program to use for remote visits depends upon whether protected health information is being discussed as part of the remote visit. If that is the case, the option would be the UVMMC HN Zoom option using a uvmhealth.org ID. It is possible to use the LCOM Zoom option, however there are specific settings and processes required as detailed below.
If no PHI is anticipated, then Teams would be an appropriate option.
Once a program has been chosen Zoom vs Teams, the option chosen must be used throughout the life of the protocol unless the protocol is amended.
Settings/Process for LCOM Zoom License
- Ensure that the host and all cohosts are using the most updated version of Zoom.
- Encourage participants to update to the latest version of Zoom prior to joining the meeting.
- Always use a password for meetings to prevent unwanted participants from entering.
- Use the Waiting Room to control access to the meeting. This allows the host to vet participants before providing access to the actual meeting.
- When all participants have joined enable the “Lock Meeting” feature to prevent additional participants from joining.
- Control the ability for participants to speak by muting them and preventing them from unmuting themselves. Unmute them as necessary to allow participation. If a participant is disruptive or their content is inappropriate, immediately mute them, eject them from the meeting, or place them back in the “waiting room”.
- Prevent participants from screen sharing unless doing so is a required component for the type of meeting that is being held.
- Assign co-hosts to manage Zoom to allow presenter to focus on presentation.
- Ensure that hosts and co-hosts know how to stop video for any participant should the need arise.
- Test the scheduling and management of meetings in advance using the locations and hardware that will be used in the real meeting.
Software to be used for eConsent
- Redcap –FDA compliant and HIPAA compliant - can be used for eConsent when there is PHI.
- Qualtrics – Neither FDA compliant nor HIPAA compliant - can be used for eConsent when there is no PHI.
Determining Which Software to Use for eConsent
The researcher needs to identify the appropriate software to use for their protocol based upon the needs of the project and what is allowed as described above. Redcap is better suited to clinical research protocols whereas Qualtrics is better suited to non-clinical studies.
Once a software has been chosen, RedCap vs Qualtrics, the option chosen must be used through the life of the protocol unless the protocol is amended.
There are two options when building eConsent in either REDCap or Qualtrics. Both products are survey tools. So, the consent would be developed utilizing fields in a database. The build may be images of each of the IRB-approved pages inserted in fields or the build may be consent text copied from the IRB-approved consent form into fields. The pros and cons of these two options are outlined in section 9.9 Electronic Consent. Links to guidance on building eConsent are below.
- REDCap eConsent build using consent page images can be found on the LCOM Commons site here.
- REDCap eConsent build using copied text can be found below.
- Qualtrics eConsent build guidance can be found on the Qualtrics site here.
- Other information about Qualtrics can be found here.
Database Software for eData Collection and Storage
- Redcap –FDA compliant and HIPAA compliant - can be used for data capture that includes PHI.
- Qualtrics – Neither FDA compliant nor HIPAA compliant - can be used for data capture when there is no PHI included.
Determining Which Software to Use for Data Collection and Storage
The researcher needs to identify the appropriate software to use for their protocol based upon the needs of the project and what is allowed as described above. Redcap is better suited to clinical research protocols whereas Qualtrics is better suited to non-clinical studies.
Once a software has been chosen, RedCap vs Qualtrics, the option chosen must be used through the life of the protocol unless the protocol is amended.
Best Practices for Communications with Participants
When utilizing email, please include this disclaimer and warning in all emails:
DISCLAIMER: This e-mail is intended only for the individual to whom it is addressed. It may be used only in accordance with applicable laws. If you received this e-mail by mistake, notify the sender and destroy the e-mail.
WARNING: E-mail sent over the Internet is not secure. Information sent by e-mail may not remain confidential.
If the message includes PHI, use secure email or inform participants of the risks associated with using unsecure email before sending electronic communications and confirm participants agree to receive communications by email.
Text Messaging
If the message sent is not PHI, it is permissible without any kind of disclaimer or warning.
If the message includes PHI, inform the participants of the risks associated with using unsecure text messaging before sending electronic communications and confirm participants agree to receive communication via text.
If messages will be sent out via autodialer, include an opt out mechanism, such as:
Reply “STOP” If you do not wish to receive additional communications via text.
Ensuring Participation After Consent
Often, individuals will not pick up a phone call or respond to a text if they do not recognize the phone number of the caller. When first recruited and consented for a study, researchers should ask participants to add the study contact’s phone number in their phone contact list at that time. The number should be one that is owned by UVM and not a personal phone number for a researcher or research team member.
Protecting Data Collected Using Remote Platforms
Data collected via remote mechanism, including recorded calls and videos, are personally identifiable and need to be protected. Given the research purpose of remote data collection, it is likely that the content often includes information that is either PII, PHI, privileged, or proprietary. You must have this addressed in your Data Security and Management plan form.
Verifying Participant’s Identity
When consent is obtained remotely, the web-form sent to the subject/subject’s legally authorized representative is not entirely secure in the sense that anyone who has access to the particular unique link for that individual can enter data and submit signatures without verifying their identity. There are a few options for ensuring additional real-time identity verification prior to eConsent. Each revolves around requesting the subject/subject’s legally authorized representative enter a passcode/information, which is established with the study staff outside of electronic communication. This can occur during an in-person meeting, but typically will occur through a telephone or Zoom or Teams call, or through information already known by both parties.
Verification with an Established Passcode: In this approach, an agreed passcode is communicated between the subject/subject’s legally authorized representative and the study team. This passcode is saved as part of the subject’s record for verification use later. The subject/subject’s legally authorized representative at the time of accessing the survey/eConsent must then enter the passcode which is compared with the stored version entered by the study staff.
Verification with Known Information: In this approach, the study team can choose to simply add questions to answer at the time of accessing the survey/eConsent. These questions should be pre-established security questions such as “What is your favorite color?” or “What is the name of the street you grew up on?” that are included on the signature page of the eConsent. The responses should be agreed upon by both the study team and the subject/subject’s legally authorized representative during an in person or during a remote (video or telephone) conversation. The answers will be saved as part of the subject’s research record for verification use later.
Verification with a Passcode Based on Known Information: In this approach, a study team who has collected sufficient demographic data can verify authentication without agreeing to a prior known passcode by simply informing the subject/subject’s legally authorized representative that a combination of their demographic data will be used as their passcode. This is a more robust form of authentication in the sense that no transmission of information between the subject and study team is required, because the information should already be known by both. For example, when obtaining eConsent a study coordinator who has collected or has access to the subject’s date of birth, middle name, and street name may choose a combination of these variables to represent the passcode, which the subject/subject’s legally authorized representative would then be prompted to answer when accessing the eConsent.
REDCap build instructions using text (borrowed from Duke University School of Medicine)
- Create a form including the consent language
- This form must be enabled as a survey
- Add descriptive fields with the consent language
- Add a field, a radio button with choices applicable to the project (i.e. I have read the consent document and I wish to participate in the study and I have read the consent document and I DO NOT wish to participate in the study, etc.)
- ONLY if participant consents should the fields below be available (branching logic must be setup)
- Use Stop Actions option when participant declines consent
- Add field to collect first name
- Add field to collect last name
- Add field to collect ‘date of birth’ (for some studies)
- Add field to collect participant’s email (be sure to validate this field as an e-mail field)
- Add field to collect signature (signature can be drawn with mouse, stylus, or finger)
- Add field to collect the date/time
- Use Action Tags @NOW to default date/time and @READONLY to make field un- editable
- Add field which includes consent version date
- Use Action Tags @DEFAULT to default date and @READONLY to make field un-editable
- Add field which includes consent expiration date
- Use Action Tags @DEFAULT to default date and @READONLY to make field un-editable
- Add Descriptive text (with optional Image/Video/Audio/File Attachment) so participant can download a copy of the unsigned consent (optional)
Survey Confirmation
To send an automatic confirmation email with the signed PDF consent document attached, follow the steps below.
- Click on ‘Project Setup’
- Under ‘Main Project Settings’, click on ‘enable’ to use surveys in the project.
- Click on the Online Designer
- Scroll down the page to the eConsent Framework section
- Select Auto-Archiver + eConsent Framework
- Under ‘Force signature field(s) to be erased if participant clicks Previous Page button while on the certification page?’, select the signature field for Signature field #1:
- Select another signature field as needed if additional signatures are collected
- Set “Send confirmation email” to ‘Yes’
- Customize the email message - Use an email address for the study team
- Check ‘Include PDF of completed survey as attachment’
- Test to verify this is working correctly (i.e., copy of signed consent is automatically saved to the File Repository)
10. Data Protection Regulations
10.1 HIPAA
HIPAA refers to the Health Insurance and Portability Act of 1996. It is a broad federal law, only part of which is intended to protect the privacy of healthcare information. It is divided into three parts: portability, accountability, and administrative simplification. There are several sets of HIPAA regulations. The most important regulation under HIPAA for research are the privacy regulations, often referred to as the Privacy Rule. The intent of the Privacy Rule is to protect the private individual’s health care information. It defines the means by which individuals/human research subjects can be informed of how their health information will be used or disclosed and it gives individuals a number of rights with regard to their health information. This information is contained in the medical consent template. The consent/authorization must be signed by the subject prior to beginning any research activities. See our HIPAA Research FAQs for more information.
The HIPAA Privacy Rule comes into play when you are conducting research within a covered entity or when you are receiving identifiable healthcare information directly from a covered entity. A covered entity is any institution that is providing healthcare to individuals, such as UVM Medical Center. If your protocol is being conducted at UVM Medical Center or you are receiving healthcare information from UVM Medical Center, HIPAA materials will be required. In some cases, when consent is waived or documentation of consent is waived, HIPAA may also be waived or altered (waiving written authorization), see above, Applying for a Waiver of Documentation, an Alteration or a Waiver of Consent.
10.2 Waiver, Partial Waiver or Alteration of HIPAA Authorization 45 CFR 164.512
UVM Medical Center and the UVM LUSE Center may use or disclose PHI for research purposes without patient authorization if the IRB (functioning as the Privacy Board) has approved a “Waiver of Authorization.”
In order to use or disclose PHI with a waiver of authorization, the IRB or Privacy Board must find:
(a) The use or disclosure of PHI involves no more than minimal risk to the privacy of individuals, based on, at least, the presence of the following criteria:
(1) An adequate plan to protect the identifiers from improper use and disclosure;
(2) An adequate plan to destroy the identifiers at the earliest opportunity consistent with conduct of the research, unless there is a health or research justification for retaining the identifiers or such retention is otherwise required by law;
(3) Adequate written assurances that the PHI will not be reused or disclosed to any other person or entity, except as required by law, for authorized oversight of the research study, or for other research for which the use or disclosure of PHI would be permitted under the HIPAA privacy rule.
(b) The research could not practicably be conducted without the alteration or waiver; and
(c) The research could not practicably be conducted without access to and use of the protected health information.
An alteration of HIPAA Authorization may be granted when there is also a request to waive the documentation of consent.
To apply for a waiver or alteration of authorization for research purposes, the Principal Investigator must complete the appropriate eform sections in UVMClick-IRB and submit it to the IRB with the initial submission documents. The IRB will evaluate the request to ensure that the waiver criteria set forth above are met. If granted, the waiver is approved by the IRB chair or designee and the Principal Investigator will be notified.
Note: List of PHI Identifiers That Make Health Information Identified
1. Names.
2. All geographic subdivisions smaller than a state, including street address, city, county, precinct, ZIP Code, and their equivalent geocodes, except for the initial three digits of a ZIP Code if, according to the current publicly available data from the Bureau of the Census:
- The geographic unit formed by combining all ZIP Codes with the same three initial digits contains more than 20,000 people, and the initial three digits of a ZIP Code for all such geographic units containing 20,000 or fewer people are changed to 000.
3. All elements of dates (except year) for dates directly related to an individual, including birth date, admission date, discharge date, date of death; and all ages over 89 and all elements of dates (including year) indicative of such age, except that such ages and elements may be aggregated into a single category of age 90 or older. The population of a zip code can be identified on the web site of the U.S. Census Bureau at the following url: http://www.census.gov/popfinder/
4. Telephone numbers.
5. Facsimile numbers.
6. Electronic mail addresses.
7. Social security numbers.
8. Medical record numbers.
9. Health insurance plan beneficiary numbers.
10. Account numbers.
11. Certificate/license numbers.
12. Vehicle identifiers and serial numbers, including license plate numbers.
13. Device identifiers and serial numbers.
14. Web universal resource locators (URLs).
15. Internet protocol (IP) address numbers.
16. Biometric identifiers, including fingerprints and voiceprints.
17. Full-face photographic images and any comparable images.
18. Any other unique identifying number, characteristic, or code, unless permitted by the individual
Request for a Partial Waiver of Authorization for Recruitment Purposes
Recruitment of potential research subjects at UVM Medical Center/UVM is a regulated research activity and must be conducted in accordance with the policies and procedures promulgated by the UVM Institutional Review Board.
A UVM Medical Center Health Care Provider or his/her agent may, without patient authorization, review the medical records of patients with whom he or she has a current clinical relationship to determine whether they meet the eligibility criteria for enrollment into a research study, and then contact prospective subjects directly by telephone or by letter, explaining the research study and requesting a decision concerning the individual’s potential interest in the study.
A “UVM Medical Center Health Care Provider” is defined as a licensed health care professional who is employed on a full-time or part-time basis by UVM Medical Center, regardless of whether or not the provider holds a faculty appointment at UVM or has an employment relationship with UVM. This includes the provider’s immediate practice group or coverage group.
A “current clinical relationship” shall be deemed to exist whenever the patient, at the time the recruitment activity is taking place, is considered to be under the care of the provider engaged in the recruitment activity or a member of the provider’s immediate practice group or coverage group.
An “agent” is defined as an individual who is under the direct supervision and control of the UVM Medical Center Health Care Provider engaged in the recruitment activity or under the direct supervision and control of a member of the provider’s immediate practice group or coverage group.
In all other cases, UVM Medical Center may only use or disclose PHI for recruitment purposes if the use or disclosure has been authorized by the patient or the researcher has obtained an IRB waiver of authorization. To obtain a partial waiver for recruitment purposes, complete the appropriate eform sections in UVMClick-IRB and submit to the IRB at the time of initial submission.
10.3 European Union (EU) Participants and EU General Data Protections (GDPR)
The European Union’s General Data Protection Regulation (EU GDPR) regulates the use, access, collection, and processing of all personal data from the European Economic Area (EEA). The GDPR apply in the 28 member states of the EU and the three additional countries (Iceland, Liechtenstein, and Norway).
Data to which GDPR applies is broader than that covered under the Health Insurance Portability and Accountability Act of 1996 (HIPAA). It applies to all personal data across all sectors of the economy, not only health care; there is no concept of a covered entity.
GDPR applies if an organization or individual is established in the EEA and acts as a data controller or processor (defined below), the organization or individual offers goods or services to individuals in the EEA, or an organization or individual monitors the behavior of individuals (multisite research or mobile application research) in the EEA.
GDPR Definitions
Controller (sponsor): the controller determines alone or jointly with others the purposes and means of processing personal data. Regulated under GDPR. Controllers provide notices to data subjects, responds to subject rights, appoints representative in EEA, notifies supervisory authorities and data subjects of data breaches, and maintains records of processing.
Processor: Processes personal data on behalf of the controller. Regulated under GDPR.
Data Subject: means an identifiable natural person who is either a citizen of the EU country (whether physically in the EU or in the US) or a US citizen who is physically in the EU for which we collect or process personal data.
EEA Countries: consist of the following: Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden and the United Kingdom. Please note the UK is still part of the EU.
Identifiable Natural Person: means a person (not an organization or company) who can be identified, directly or indirectly, in particular by reference to an identifier.
Identifier: includes data elements such as a name, an identification number, location data, an online identifier or to one or more factors specific to the physical, physiological, genetic, mental, economic, cultural or social identity of that natural person.
Personal Data: under GDPR means any information that can be used to identify a natural person that relates to an identified or identifiable nature person (data subject) regardless of the medium in which is exists (i.e., paper, electronic, recorded, video, audio) and regardless of how/where it is stored (i.e., server, laptop, thumb drive, on the cloud, paper files in a file cabinet, etc.)
How could GDPR impact research at UVM
1. If you are collecting or will collect “identifiable personal data” from participants residing in the EEA on or after May 25, 2018, your project will be subject to GDPR.
If a PI at UVM/UVMMC is the sponsor of a multi-site, international protocol that involves one of these countries, GDPR applies because data is being exchanged and services are being provided through a Clinical Trial Agreement between the US-based investigator (sponsor) and the EEA study site. In this instance, the UVM PI must ensure that appropriate notice about the GDPR is provided to those EEA subjects. Since the single IRB requirement does not exist for international protocols, local EEA IRB review would be required. It is assumed that the notice to EEA subjects will be enforced by the local IRB. However, the UVM PI must be aware and understand this requirement.
2. If a UVM/UVMMC investigator conducts research in the U.S. and transmits identifiable personal data to sponsors, servers, or data core facilities within the EEA on or after May 25, 2018, your project will be subject to GDPR.
In this instance, the sponsor organization is established in the EEA and acts as a data controller or processor. Sponsor is offering goods or services by virtue of a Clinical Trial Agreement. The sponsor of the protocol will be responsible to provide their notice to the UVM researcher for distribution to subjects. The privacy notice must include contact information for the sponsor in the event that a subject wishes to apply any of their data privacy rights. It is not appropriate to list the local privacy officer as he/she does not administer the EU GDPR. The requirement to include a signature of the subject on the notice is at the sponsor’s discretion. The IRB does not require signature on the privacy notice. The sponsor’s notice and any script used to explain this regulation to subjects in the US is to be submitted to the IRB for review.
3. UVM student projects collecting identifiable personal data from EEA subjects on or after May 25, 2018 may be subject to GDPR.
It is not clear that UVM student projects would be providing any goods or services, typically these are survey projects and there is no Clinical Trial Agreement. Contact the office in these instances.
4. UVM mobile research studies that collect personal data from EEA residents may also be subject to the GDPR.
For instance, US-based entity provides mobile application to EEA residents for collection of research data. UVM/UVMMC is offering good or services but there may not be a Clinical Trial Agreement. Contact the office in these instances.
5. European governmental grants or contracts may require compliance with GDPR.
Personal data flows with EEA direct grant awardees or sub-recipients should be scrutinized to see if they involve offering services to EEA data subjects. Contact SPA or RPO to assist in these instances.
What if I am not collecting personal data from individuals in the EEA?
In short, GDPR would not apply. Research studies may not involve the receipt of personal data because the data received may not relate to an identified or identifiable natural person. For example, studies that do not collect information that is linked to a subject’s identity, such as anonymous surveys in which the identities of survey subjects cannot be traced, would not involve the receipt of personal data.
What if I am only receiving coded data?
The GDPR considers key-coded data to be “personal data” and refers to key-coded data as “pseudonymized data”. This is in contrast to the position under many U.S. research and privacy laws, such as the Common Rule and HIPAA. Pseudonymized data are regarded as identifiable personal data and therefore remain subject to the GDPR’s protections, even when in the hands of a person who lacks the key needed to link the data to the data subject’s identity.
Is it possible to de-identify data so that GDPR does not apply?
The GDPR does not apply to data that have been “anonymized.” However, for data to be anonymized, the GDPR requires that there be no key to re-identify the data. For example, if UVM serves as the sponsor of a research study with a site located in the EEA and receives only key-coded information from the EEA site, the key-coded data from the EEA site remain “personal data” in the hands of UVM. This is the case even if UVM has no access to the key needed to re-identify the coded data. Unlike in HIPAA, there is no “safe harbor” under the GDPR to which data can be rendered de-identified by removing a specific list of identifiers. Rather, anonymization is judged on a facts and circumstances basis taking into account all the means reasonably likely to be used, such as singling out, either by the controller or by another person to identify the natural person directly or indirectly. Given this definition, anonymization is an extremely high standard that is difficult to meet in practice.
I have heard that subjects have additional rights under the GDPR. Is that true?
Yes, it is. The GDPR creates a range of rights that are available to research subjects under certain situations. Some of the rights under the GDPR include the right that research subjects can obtain copies of all of their personal data and have the right to withdraw consent to further processing of their personal data. Upon withdrawal of consent for research, one can no longer retain the personal data for the purpose of research, including in pseudonymized (key-coded) form. However, one may retain the data if necessary for legal compliance (i.e., for adverse event reporting). Also, the researcher could continue to process the data for research purposes if the data have been fully anonymized through removal of all identifiers associated with the data, including destruction of the key linking the subject’s data to his or her identity (Please see previous note on “anonymized” data).
10.4 Decedent Data
While the Common Rule does not apply to decedents (and thus does not require IRB review), the Privacy Rule does apply. Specifically, the Privacy Rule requires researchers to provide written assurance that they will protect the privacy of decedents' identifiable health information. Important to remember is that the Privacy Rule does not distinguish between living and deceased subjects in terms of the requirement for tracking disclosures pursuant to a waiver of authorization. Therefore, tracking of disclosures of decedents' PHI will be required by the investigator. You may find the UVMMC Attestation Form for Decedent Research in the Forms Library.
10.5 National Institutes of Health Genomic Data Sharing Policy
New 09/01/21
The National Institutes of Health (NIH) Genomic Data Sharing (GDS) Policy, effective January 25, 2015, sets forth expectations that ensure the broad and responsible sharing of genomic research data in proposals. Frequently asked questions can be found here.
Scope
The GDS Policy applies to all NIH-funded research that generate large-scale human or non-human genomic data as well as the use of these data for subsequent research. NIH expects all funded investigators to adhere to the GDS Policy.
Adherence to the data sharing plan, as outlined in the policy, for non-NIH funded genomic data is not required but encouraged.
Investigator Responsibilities
Investigators who intend to use research or clinical specimens collected or cell lines created after January 25, 2015 to generate genomic data, may only do so when informed consent processes explicitly discuss future research use and broad data sharing, even if the data are generated from specimens that are de-identified. NIH-designated data repositories (database of Genotypes and Phenotypes (dbGaP) will not accept genomic data after this date without this type of consent.
Specific guidance about study registration and data submission to a NIH-Designated Repository can be found at this link. There is specific guidance for investigators regarding the language to be used in the consent form. The IRB will be making its standard determinations when reviewing these projects but will also ensure that the consent form includes the required elements which can be found at the National Human Genome Research Institute.
Investigators seeking NIH funding should be in communication with their Program Official as early as possible to discuss data sharing expectations and timelines that would apply to their proposed studies. NIH expects investigators and their institutions to provide basic plans to follow this policy.
An Institutional Certification (for sharing human data) will be required as part of the terms and conditions of funding. This certification is completed by the investigator and signed by the Institutional Official or his/her designee. The Director of the Research Protections Office will sign these certifications once the IRB has met it responsibilities as outlined below.
Institutional Responsibilities
Investigators should work with their Research Administrator within UVM’s Sponsored Projects Administration to ensure completion and signature on the Institutional Certification form.
The certification assures that:
- The data submission is consistent with all applicable laws and regulations, as well as institutional policies;
- The limitations on the research use of the data, as expressed in the informed consent documents, are delineated;
- The identities of research participants will not be disclosed to the NIH-designated data repositories;
- IRB reviewed and made its determinations (below);
- It is clearly indicated as to whether the data are to be made available through unrestricted or controlled-access and whether future use is limited to not-for-profit organizations.
Additionally, any applicable institutional agreements should be in place to share this data with the NIH either through Sponsored Programs, Office for Clinical Trials Research, or the Data Management Office at UVMMC.
IRB Responsibilities
As outlined by the GDS Policy, the IRB is responsible for determining the following:
- The protocol for the collection of genomic and phenotypic data is consistent with human subject regulations;
- Data submission and subsequent data sharing for research purposes are consistent with the informed consent of study participants from whom the data were obtained;
- The investigator’s plan for de-identifying datasets is consistent with the standards outlined in the GDS policy; and
- To the extent relevant and possible, consideration was given to risks to groups or populations associated with submitting data to NIH-designated data repositories and subsequent sharing;
- For retrospective data, if the consent under which the existing genetic materials and data were obtained, is consistent with the submission of data to the NIH and the sharing of data in accord with the GDS policy.
For studies that propose to use existing data or samples, the IRB may be forced to conclude that the original consent form is not adequate for submission to the NIH repository. In these cases, the IRB may decide that it is appropriate and necessary for the investigator to seek explicit consent of the research participants.
If the IRB has granted a waiver of some or all the required elements of informed consent under the relevant provisions of 45 CFR 46.116, or if consent is not required because the activity is not subject to 45 CFR 46, investigators are required to seek, and document consent for future use and broad sharing of genomic and phenotypic data to meet NIH expectations under the GDS Policy.
The Policy goes beyond the regulatory requirements because genotype and phenotype information generated about individuals will be substantial and, in some instances, sensitive (such as data related to the presence or risk of developing specific diseases or conditions and information regarding family relationships or ancestry), the confidentiality of the data and the privacy of participants must be protected.
11. Certificates of Confidentiality
Revised 10/4/2023
Purpose
Certificates of Confidentiality (CoCs) protect the privacy of research subjects by prohibiting disclosure of identifiable, sensitive research information to anyone not connected to the research except when the subject consents or in a few other specific situations.
Scope
The NIH policy applies to all biomedical, behavioral, clinical, or other research funded wholly or in part by the NIH whether supported through grants, cooperative agreements, contracts, other transactions awards, or conducted by the NIH Intramural Research Program, that collects or uses identifiable, sensitive information.
The NIH will continue to consider request for CoCs for non-federally funded research in which identifiable, sensitive information is collected or used.
Any projects with CoCs issued in the past, regardless of funding sources, must comply with the requirements of the CoC policy, especially the new disclosure requirements and restrictions.
• The new disclosure requirements prohibit disclosure of the name of research subjects or any identifiable research information, document, or biospecimen to anyone not connected with the research except under very specific circumstances as detailed in the CoC policy.
Who Issues CoCs
Who issues CoCs depends upon who is funding the research.
NIH Funded
Effective October 1, 2017, NIH will automatically issue CoCs to all research funded by NIH that is collecting or using identifiable, sensitive information. NIH funded researchers are automatically issued a CoC through their award. Separate documentation of the CoC will not be provided by NIH.
Other HHS Agencies (Non-NIH)
Several non-NIH HHS agencies, including CDC, FDA, BARDA, IHS, HRSA, and SAMHSA, issue CoCs.
Non-HHS Federal Agencies
If your research is funded by a non-HHS Federal Agency other than HHS, you may request a CoC for a specific project that involves sensitive, identifiable information.
Non-Federally Funded Research
Health-related research that is not federally funded in which identifiable, sensitive information is collected or used, may request a CoC for specific projects.
Extent and Limitations of Coverage
Disclosures are only permitted when:
• Required by Federal, State, or local laws (e.g., as required by the Federal Food, Drug, and Cosmetic Act, or state laws requiring the reporting of communicable diseases to State and local health departments), excluding instances of disclosure in any Federal, State, or local civil, criminal, administrative, legislative, or other proceeding;
• Necessary for the medical treatment of the individual to whom the information, document, or biospecimen pertains and made with the consent of such individual;
• Made with the consent of the individual to whom the information, document, or biospecimen pertains; or
• Made for the purposes of other scientific research that is in compliance with applicable Federal regulations governing the protection of human subjects in research.
Limitations on Issuance of CoCs for Data and Biospecimen Repositories
For research not funded by NIH, issuance of a COCs are limited to single projects. CoC requests for non-NIH funded research that involve multiple projects, studies, or protocols or that include a research program, will not be approved.
• For research not funded by NIH, CoC requests for the establishment and maintenance of a data or biospecimen repository that include plans to conduct studies and projects with those data and/or biospecimens are considered research programs and will not be approved.
• NIH will consider CoC requests for research not funded by NIH for an individual study that will use data or biospecimens from a repository.
• NIH also limits CoCs to NIH-funded repository studies. CoCs may be issued for the establishment and maintenance of research repositories where the main source of the data and/or biospecimens was originally obtained for clinical care or other purposes, rather than research purposes.
• CoC requests for research not funded by NIH (e.g., discretionary CoCs) for repositories where the main source of the data and/or biospecimens was originally obtained for clinical care or other purposes will not be approved.
Applying for a CoC when the Project is Not NIH Funded
Please note, an application for a CoC should be submitted before the IRB reviews the protocol
1. Determine if a CoC is Appropriate
Research projects that collect personally identifiable, sensitive information are eligible for a CoC. Requesting sensitive information from a participant does not automatically make it eligible for a CoC. The law focuses on the identifiability of the information.
Some research areas that are eligible for a CoC are:
• Research on HIV, AIDS, and other STDs;
• Studies that collect information on sexual attitudes, preferences, or practices;
• Studies on the use of alcohol, drugs or other addictive products;
• Studies that collect information on illegal conduct;
• Studies that gather information that if released could be damaging to a participant’s financial standing, employability, or reputation within the community;
• Research involving information that might lead to social stigmatization or discrimination if it were disclosed;
• Research on participant’s psychological well-being or mental health;
• Genetic studies, including those that collect and store biological samples for future use;
• Research on behavioral interventions and epidemiologic studies.
Projects that are not eligible for a certificate are:
• not research based;
• not collecting sensitive information or information that, if released publicly, might harm the research participants,
• not collecting personally identifiable information, or
• not involving a subject matter that is within a mission area of the National Institutes of Health.
2. Applying for a Certificate
If your research is funded by an HHS agency other than NIH, such as BARDA, CDC, FDA, IHS, HRSA or SAMHSA or your research is funded by a non-HHS agency or the research is not federally funded, you still may request a CoC for specific health-related projects using sensitive, identifiable information. If your study is federally funded by one of the above listed agencies, you should contact the CoC Coordinator at your funding agency for questions on how to obtain a CoC.
Investigators whose research is not NIH-funded and is not funded by the above-listed agencies/departments should use the online NIH Certificate of Confidentiality application system to request a CoC. Issuance of a CoC will be at the discretion of NIH.
Note: NIH will not issue a CoC for Agency for Healthcare Research & Quality (AHRQ) or Department of Justice (DoJ) funded research. Contact AHRQ and DoJ respectively, to obtain information about their privacy regulations.
Certificates are granted to institutions (not investigators nor IRBs), based upon an investigator’s application, for single, well-defined research project. Certificates are sometimes issued for cooperative multi-site projects which must have a coordinating center or lead institution. The coordinating center/lead institution can apply on behalf of all institutions associated with the multi-site project and must ensure that all participating institutions conform to the application assurances.
As part of the application, a CoC Assurance must be signed by UVM’s authorized institutional official. The UVM Board has delegated authority to the Executive Director for Research Administration. The CoC system sends an auto-generated email to the institutional official listed on the CoC request with a hyperlink to the CoC request. The institutional official needs to review the CoC request information for accuracy and affirm the online Institutional Assurance Statement by checking each box and then submitting the CoC request.
NIH-Funded Research
CoCs automatically cover research activities and do not need to be extended or amended while the research remains funded by NIH. CoC protections also continue for the duration of a no-cost extension. If your NIH-funding will or has ended but the collection of new data from research participants will continue without NIH-funding, you will need to apply for a CoC for continuity of protections using the CoC application system. If your NIH funding will or has ended but your study has completed all enrollment and data collection, there is no need to extend the Certificate. Sensitive, identifiable research information maintained by investigators during any time a Certificate is in effect, is protected permanently.
IRB Review
The informed consent form must include a description of the protections and limitations of the CoC in the confidentiality section, including the circumstances in which the investigators plan to disclose voluntarily identifying information about research subjects (see Section 12 -Exceptions to Confidentiality Guidance). Consent language for this section can be found within the consent template found on the IRB forms page.
The protocol should include the fact that the study will have a CoC. If during its review of research for which an investigator has not identified the need for a CoC, the IRB may recommend applying for a CoC. The IRB may hold approval of the study until the CoC has been obtained and is on file with the IRB.
Certificate Expirations/Extensions
CoC protections remain in perpetuity for already collected or used information. A new CoC will need to be obtained to cover any new data collected from already enrolled participants or any new participants if the non-NIH funded research activity will extend beyond the expiration date on the Certificate or if data collection will continue after NIH funding ends. You can submit a request for a new CoC through the Online CoC System. Note: NIH-issued CoCs for non-NIH funded research activities issued on or after January 12, 2021 do not have an expiration date.
If your study has completed all enrollment and data collection, there is no need to extend the Certificate. Sensitive, identifiable research information maintained by investigators during any time a Certificate is in effect, is protected permanently.
Amending a Certificate
If there is a SIGNIFICANT change in your non-NIH funded project after the Certificate has been issued, you should request a new Certificate through the NIH online CoC system. This includes such changes as a major change in the scientific scope of the project, changes in the PI, or in the drugs to be administered. For the purpose of the Certificate, minor changes, such as the addition of a new survey instrument or clinical test or adding a participating research site to a multisite study, are not considered a significant change, and do not require a new Certificate.
Application Contacts
For more information, please see the NIH COC website. You may direct CoC questions to the NIH Office of Extramural Research: NIH-CoC-Coordinator@mail.nih.gov.
12. Exceptions to Confidentiality Guidance
Mandatory Reporting
Vermont law requires health care providers and other professionals to take specific actions and to submit specified reports when certain facts or conditions become known to them (i.e. child abuse, elder abuse and imminent harm to self or others). These laws apply to the professional as a condition of their license or employment; and thus to any information gained by that person during the conduct of research.
The IRB expects that the PI will, when appropriate, communicate these potential exceptions to confidentiality to prospective research subjects during the informed consent process. This applies both to research projects specifically gathering such information and to projects where these circumstances or conditions may become known to the researcher even though the researcher does not directly seek this information (i.e. abuse of a child may be evident during a physical exam).
Do I need to include an Exceptions to Confidentiality section in my consent form?
If you are subject to mandatory reporting laws AND there is a reasonable possibility that you could discover information that would require you to break confidentiality then your consent form should include an Exceptions to Confidentiality section.
Am I (or any Key Personnel on this study) subject to mandatory reporting laws?
Health care workers, mental health providers, social workers, educators, members of the clergy, and law enforcement officers are examples of professionals who are mandated reporters in Vermont. Check with your professional licensing board if you are unsure if you are a mandated reporter. In addition, UVM requires any employee who has reasonable cause to believe that a minor participating in a program or activity at the University has been sexually abused or neglected to report the concern promptly to the Vermont Department for Children and Families (DCF).
What is the likelihood that in the course of carrying out my protocol I will discover information that requires mandatory reporting?
If the likelihood is low then the consent form does not need to include additional language. If during the conduct of the research a situation that required mandatory reporting arises, the PI should first and foremost act ethically to protect a potential victim (i.e. report suspected child abuse to the authorities). In addition the PI should report this to the IRB as the breach of confidentiality would be an Unanticipated Problem.
If there is a reasonable possibility, this should be disclosed as an exception to confidentiality to the potential participant during the informed consent process.
Suggested Consent Language
Place this consent language in the confidentiality section of the consent.
There is one exception to confidentiality that you should know about. By law, it is our responsibility to report to the appropriate authority suspicion of harm to children or to others.
If appropriate add: However, we are not seeking this type of information in our study nor will you be asked questions about these issues.
13. Cooperative Research (Single IRB) Policy (Sec. __.114)
Revised 05/10/2023
The definition of cooperative research is any research project that involves more than one institution. In the conduct of cooperative research projects, each institution is responsible for safeguarding the rights and welfare of human subjects and for complying with Cooperative Research regulations.
Cooperative Research Regulations
As of January 25, 2018, NIH policy required that all U.S. sites participating in multi-site, non-exempt, human subjects research funded by the NIH use a single Institutional Review Board (sIRB) to conduct the ethical review required for the protection of human subjects.
As of January 20, 2020, the revised Common Rule required at 45 CFR 46.114(b) that all institutions located in the United States that are engaged in cooperative research conducted or supported by a Common Rule department or agency rely upon approval by a single IRB for the portion of the research that is conducted in the United States.
Neither of these regulatory mandates applies to exempt research. Therefore, the UVM IRB will not rely on another IRB for review of exempt projects nor act as the single IRB for a multiple-site exempt project. Additionally, the UVM IRB will not rely on another IRB or act as the single IRB if it is determined that UVM/UVMHN is not engaged in the research, because the regulatory mandates do not apply to non-engaged sites. (see Section 3.7 Determination of Institutional Engagement in Research). UVM/UVMHN PIs should contact the IRB for help determining local engagement.
Single (Reviewing) IRB
Except under specific circumstances described below, the UVM IRB will not act as the IRB of record for multi-site research that falls under the NIH sIRB Mandate or Common Rule regulations. UVM has subcontracted with WCG IRB (formerly Western IRB or WIRB) for single IRB services where UVM/UVMHN researchers wish to be the lead site for their federally funded proposals. Proposals should include the use of WCG IRB as the IRB of record for the multi-site non-exempt research activities and budgets must be developed to include the expense for the use of this commercial IRB.
Relying Site
If UVM is not the lead site, UVM/UVMHN will participate and rely on another IRB for review of non-exempt projects, with appropriate reliance agreements in place. See Section 13.3. Procedures for Relying on External IRB for Federally Funded Research.
The UVM IRB will continue to review all projects that that are not federally funded, unless use of a single IRB is mandated by the lead PI or research consortium/network.
13.1 Cooperative Research (Single IRB) Roles and Responsibilities
Revised 04/20/2023
The External IRB will be responsible for regulatory review at the Expedited or Committee level and ensuring that the protocol meets the regulatory requirements for protecting human subjects. The University of Vermont and the University of Vermont Health Network (UVMHN) are responsible for local oversight of human subject protections. Each of the following roles play an important part in protecting human subjects. This list may not be all-inclusive and responsibilities may change based on the terms set forth in reliance agreements.
Relying Institution (UVM/UVMHN) Responsibilities
The following responsibilities are examples of conditions typically defined in reliance agreements.
UVM/UVMHN as the Relying Institution
Both institutions are responsible for the following, regardless of which IRB reviews the research:
• Education and training of our local investigators & research staff
• Quality assurance and post-approval monitoring of local research activities
• Conflict of Interest review and management
• Execution of institutional reliance agreements
• Conduct of institutional Ancillary Reviews
• Adherence to responsibilities as outlined in the reliance agreement
UVM IRB
• Decision to cede IRB review
• HIPAA determinations if acting as the Privacy Board; HIPAA implementation
• Local Context review of protocols to ensure compliance with state or local laws, regulations, and institutional policies; communication of local considerations to the Reviewing IRB as requested
• Continued local oversight of active External IRB studies until closure or termination
UVM/UVMHN Principal Investigator
• UVM/UVMHN PIs have the overall responsibility for the local conduct of the protocol. See Section 5.1 of the UVM IRB Policies and Procedures, Responsibilities of Principal Investigators.
• Must adhere to the policies and procedures of both the External IRB and the UVM IRB as well as ensure compliance with the regulatory determinations of the External IRB
• Scientific and/or feasibility review to ensure appropriateness of protocol for our site
• Prepare and submit site-specific materials to the Reviewing IRB or Lead Site (following UVM Local Context review) for initial approval
• Ongoing submission requirements to the Reviewing IRB or Lead Site including modifications, continuing reviews, reportable new information, audits, and corrective actions
• Ongoing submission requirements to the UVM IRB as detailed in Section 13.3 of the UVM IRB Policies and Procedures
• Communicate clearly and promptly with both the External IRB and UVM IRB
Designated Contact Person
The UVM/UVMHN PI needs to identify one person on their research staff as their designated contact for all interactions and communications with the External IRB. This person will work as the liaison between the other institutional point of contact or directly with the External IRB and the local PI as well as the intermediary between the External IRB and UVM’s IRB.
Each External IRB will be different as to what they require for information from UVM. The Designated Contact Person should work with the External IRB in consultation with the UVM IRB on providing the information requested. If the External IRB requests UVM’s local context information, the Designated Contact Person would provide the following two items in response:
• UVM Local Context document
• UVM Consent/HIPAA Checklist for Required Language
External IRBs may request that the Designated Contact complete a Site Information form and/or a Communications & Responsibilities document. These forms should be completed with the assistance of the UVM IRB. Please reach out to the UVM IRB Reliance Administrator for assistance.
Others
• PIs who will conduct protocols utilizing UVMHN resources will work with the UVMHN Compliance Office to create a billing plan.
• UVM Office of General Counsel will be involved with reliance agreement negotiations as necessary.
• HIPAA issues may require consultation with the UVMHN Manager of Compliance and Privacy.
• Other Institutional Committees and Departments (Radiation Safety Committee, Investigational Drug Services, Office of Clinical Trials Research, Sponsored Programs Administration, etc)
• Other departments or individuals may need to be consulted.
External (Reviewing) IRB and Lead Site’s Responsibilities
The following responsibilities are examples of conditions typically defined in reliance agreements for the lead site’s study team and institution and the Reviewing IRB.
External Lead Site
• The lead site and lead PI are responsible for the overall conduct of the research protocol across study sites.
• Ensuring study teams at relying sites are qualified to conduct the proposed research
• Preparing and submitting study materials to the Reviewing IRB for initial study review, site review, continuing reviews, modifications, and reportable new information
• Providing sites with the sIRB-approved protocol and study document templates
• Communication of Reviewing IRB determinations to sites
• Communicating study closures to participating sites and the Reviewing IRB
• Adherence to responsibilities as outlined in the reliance agreement
Reviewing IRB (External IRB, IRB of Record, Lead IRB)
• Decision to act as Reviewing IRB
• Make available to Relying Institutions the Reviewing IRB’s policies and procedures
• Conduct initial and continuing reviews of submitted research, including amendments and reportable new information, in accordance with human subjects protection regulations, ethical principles, and local requirements of the participating sites
• HIPAA determinations if acting as the Privacy Board
• Consideration of Conflict of Interest management plans from participating sites
• Continued oversight of active studies until closure or termination including quality assurance post-approval monitoring and audits
• Reporting to federal agencies and sponsors
• Prompt notification of Overall PI, Site Investigators, and Relying Institution of determinations, lapses in approval, unanticipated problems, injuries, complaints, serious or continuing non-compliance, audits, federal reporting, and corrective actions.
• Adherence to responsibilities as outlined in the reliance agreement
13.2 Collaborative Agreements
Revised 10/11/2023
DATA USE AGREEMENT
Data Use Agreements (DUAs) are contractual documents used for the transfer of non-public data (also specimens with data attached) that is subject to some restrictions on its use. DUAs serve to outline the terms and conditions of the transfer. Specifically, DUAs address important issues such as limitations on use of the data, obligations to safeguard the data, liability for harm arising from the use of the data, publication, and privacy rights associated with confidential or protected data. The understanding established by a DUA can help avoid later issues by clearly setting forth the expectations of the provider and recipient.
The use of DUAs may apply to any project where data is being shared including those projects researchers self-determine to be Projects Not Requiring IRB Review. Note: Limited data sets may also require a DUA prior to sharing.
Researchers who intend to share non-public data outside of the institution must contact either Sponsored Project Administration or the Office of Clinical Trials Research for further information regarding negotiation of a Data Use Agreement. For UVM agreements, if any of the data is Protected Health Information (PHI) from the UVM Health Network, UVMHN must also be a signatory on the Data Use Agreement. The UVMHN signing official will either be from the Office of Clinical Trials Research or the UVMHN Data Management Office, depending on the source of the PHI.
CLINICAL TRIAL AGREEMENT (CTA)
Clinical trial agreements (CTAs) are the legally binding agreements between a sponsor and an institution (site) as to how certain business and property rights will be dealt with between the parties.
SPONSORED RESEARCH AGREEMENT
A sponsored research agreement is a contract between UVM and a sponsor for the purposes of funding and conducting research at the University. A sponsored research agreement may be supported by funding from for-profit (e.g. private industry) or non-profit (state or federal government, foundations, etc.) sponsors.
INDIVIDUAL INVESTIGATOR AGREEMENT (IIA)
A formal, written, binding agreement in which UVM agrees to extend its Federalwide Assurance (FWA) approval to cover a non-assured institution or individual for the purposes of collaborating with a UVM researcher. The IIA sets out terms and conditions for the institutions/individuals. See Section 13.7 Collaborations with Community Partners.
INSTITUTIONAL SUPPORT LETTER (E.G., SCHOOLS, NURSING HOMES)
A letter signed by an executive director, chief executive officer, board president, superintendent, or other individual with authority to commit the institution’s resources, acknowledging the proposed research activity, and granting permission for the engagement of their employee and facilities (if applicable) in that activity. A template of our support letter is located on our Forms Library page.
MATERIAL TRANSFER AGREEMENT
If a researcher wishes to share biological materials collected at the University of Vermont with colleagues at another institution, the human biological materials must be transferred pursuant to a University Material Transfer Agreement (MTA) executed by UVM Innovations.
If a researcher wishes to share biological materials collected at UVM Medical Center with colleagues at another institution, the human biological materials must be transferred pursuant to a UVM Medical Center Material Transfer Agreement (MTA) executed by the Office of Clinical Trials Research.
MEMORANDUM OF UNDERSTANDING (MOU)
Researchers often collaborate and share research tools with other scientists or institutions without receiving funding. For many of these collaborations, a written agreement is beneficial or necessary. These agreements set out expectations, terms, and requirements that protect the interests of the investigators and the participating organizations. UVM investigators proposing to include an outside entity in their research should review Sponsored Project Administration’s agreement page for further information regarding negotiation of a MOU.
RELIANCE AGREEMENT
A reliance agreement (also called an IRB Authorization Agreement or IAA) is a document signed by two or more institutions engaged in human subjects research that permit one or more institutions to cede review to another IRB. The signed agreement permits a single IRB to review human subject research activities for more than one site. At this time UVM is only entering into Reliance Agreements where a UVM PI is engaged in a non-exempt, cooperative, human subjects research project that is federally funded or for which the lead PI or research consortium/network mandates use of a single IRB.
SMART IRB MASTER RELIANCE AGREEMENT
“SMART IRB (the Streamlined, Multisite, Accelerated Resources for Trials IRB Reliance platform) is designed to harmonize and streamline the IRB review process for multisite studies, while ensuring a high level of protection for research participants.”
Freely available for institutions and investigators, the SMART IRB is not a single IRB that will review collaborative research. It is an integrated, comprehensive platform that offers a Master IRB Reliance Agreement (the SMART IRB Agreement v1 and SMART IRB Agreement v2.0) and a web-based system (SMART IRB’s Online Reliance System) allowing institutions and their investigators to initiate, track, and document study-specific reliance agreements.
There are multiple participating institutions who have joined the SMART IRB. The University of Vermont IRB is an approved participant in the SMART IRB platform. This means that we have signed onto the SMART IRB Master Reliance Agreement. It is the preference of the UVM IRB to use the SMART IRB agreement as the basis of reliance for all studies where we rely on an External IRB. This agreement clearly defines the roles and responsibilities of each institution and eliminates the need to sign individual agreements for each study in which you plan to collaborate. To learn more about SMART IRB go their website at https://smartirb.org/.
StrokeNet MASTER RELIANCE AGREEMENT
“NIH has created the NIH StrokeNet to conduct small and large clinical trials and research studies to advance acute stroke treatment, stroke prevention, and recovery and rehabilitation following a stroke across the lifespan.”
UVM/UVMHN is a member of the NIH StrokeNet, which is a network of 27 regional centers across the U.S. involving approximately 500 hospitals. The network utilizes both a Central IRB at the University of Cincinnati and the commercial IRB, ADVARRA, to review StrokeNet protocols. UVM has agreed to cede IRB review to the University of Cincinnati IRB for StrokeNet protocols in which UVM/UVMHN is participating, under the SMART IRB Master Reliance Agreement.
WCG IRB MASTER RELIANCE AGREEMENT
The UVM IRB does not act as the Reviewing IRB except in certain, specific circumstances under pre-existing agreements. UVM has subcontracted with WCG IRB for Single IRB services where UVM/UVMHN researchers wish to be the lead site for their federally-funded proposals. Proposals should include the use of WCG IRB as the IRB of Record for the multi-site research activities, and budgets must be developed to include the expense for the use of this commercial IRB. Reference the UVM RPO’s WCG IRB web page for step-by-step instructions.
NATIONAL CANCER INSTITUTE CENTRAL IRB (NCI CIRB) MASTER RELIANCE AGREEMENT
For information about reliance on NCI CIRB for adult protocols contact UVM Cancer Center Clinical Trials Office. For reliance on NCI CIRB for pediatric oncology protocols, contact the Office of Clinical Trials Research.
13.3 Procedures for Reliance on External IRB (Sec __.114(b)(1))
Revised 09/21/2023
(Sec __.114(b)(1))
At this time, the University of Vermont (UVM) will allow engaged UVM and UVMHN researchers to rely on an External IRB for multicenter, domestic, non-Exempt, human subjects research protocols where the project has federal funding or single IRB is otherwise required. Currently, UVM has the following master reliance agreements in place. Master reliance agreements allow UVM to rely on a single IRB repeatedly without having to renegotiate an agreement for each individual project. UVM will document ceding review to an External IRB under an existing master reliance agreement for each individual project using a letter of acknowledgement or online reliance system, as needed.
• National Cancer Institute Central IRB (NCI CIRB) – for adult and pediatric oncology protocols
• SMART IRB – for protocols where the reviewing IRB is also a participating institution in SMART IRB
• StrokeNet – for StrokeNet protocols
• WCG IRB (formerly Western IRB/WIRB) – when UVM is the lead site for federally-funded cooperative research, UVM has subcontracted single IRB responsibilities to WCG IRB
Other types of individual or new reliance agreements will require negotiation and sign off by the Director of the Research Protections Office at UVM.
Multi-site Industry, multi-site Foundation, and multi-site internally funded protocols will continue to be reviewed by the UVM IRB, unless use of single IRB is mandated by the lead PI or research consortium/network.
UVM Study Team Steps to Allow Reliance on an External IRB
The process for establishing reliance on an External IRB can vary widely from institution to institution. UVM/UVMHN PIs are often approached by Lead Sites or External IRBs directly. Guidance should be sought from the UVM IRB early in the process to ensure the request to rely is approvable and that all necessary steps are followed. The IRB Reliance Administrator should be contacted for help executing reliance agreements and other study-specific agreements, site information and local context forms, submissions in online reliance databases, and the UVMClick submission process.
Generally, reliance will be established prior to completing the steps below. It is typically the responsibility of the Lead Site or External IRB to submit a reliance request for a specific study to a participating site. This can be done via the SMART IRB Online Reliance System, IRB Reliance Exchange (IREx), other online portals, or email. If a UVM/UVMHN PI is contacted with a reliance request, they should direct the request to the UVM IRB Reliance Administrator.
It is important that the following steps be completed in the appropriate order, to ensure the UVM IRB is able to conduct a local context review of study documents prior to UVM site submission to the External IRB.
1. Request to Allow Reliance on an External IRB
PIs must create and submit a new External IRB protocol through UVMClick. This is sometimes initiated concurrently with the reliance request from the External IRB/Lead Site. Creation of an External IRB protocol is slightly different than the process for a UVM single-site protocol. See directions here under UVMClick User Guides, “Documentation for User Community – External IRB Studies,” and required UVM forms here under IRB Forms Library “Request to Rely on Single-External IRB.”
The following documents are required for local context review at UVM:
Standard Attachments
• Protocol approved by the External IRB
• Lead Site’s template Consent/HIPAA form edited to include UVM required language (See "Consent/HIPAA Checklist for Required Language") (UVM form)
• Data Management and Security Plan (UVM form)
Additional Attachments
• A completed Request to Rely on an External IRB form (UVM form)
• The Lead Site’s template Consent document(s)
• Any reliance agreements that require UVM institutional signature
• Any local context/site information forms required by the External IRB
• The initial, study-wide approval memo from the External IRB
Additional components applicable to local context review in UVMClick may include a site-specific research plan (if local research activities differ in scope from the approved protocol), a local recruitment plan, the Sponsored Projects Administration’s Funding Proposal number (FP#) assigned to the federal grant or subaward (if external federal funds are being provided for UVM/UVMHN participation), Financial Conflict of Interest Management plan(s) if applicable, and entry of all local key personnel.
If UVM is acting as the Privacy Board or if the External IRB requires a HIPAA Authorization that is separate from the consent, there is a HIPAA Authorization template available on the Forms Library. If the External IRB is acting as the Privacy Board and issues a waiver or alteration of HIPAA Authorization, a “Documentation of Waiver or Alteration of HIPAA Authorization” may be requested as part of the local context review.
2. Local Context Review by UVM and Ancillary Departments
Appropriate, administrative local context reviews will be conducted by the UVM IRB to ensure compliance with local and state laws and regulations and institutional policies and procedures. This may involve reviews by ancillary departments and clarification requests from UVM IRB staff to the UVM Study Team through UVMClick. Additionally, if the IRB Reliance Administrator has questions or concerns during local context review that the study or its review by the External IRB may not be consistent with the local population, resources, policies, procedures or laws/regulations, there will be appropriate outreach to IRB Chairs or Designees for consultation.
Once local context review is completed, UVM IRB staff will confirm reliance in UVMClick, which will move the submission to “Pending sIRB Review” status. The UVM Study Team will be alerted that they may submit the reviewed local materials to the External IRB for regulatory approval.
Note: The Request to Rely on an External IRB form inquiries about Ancillary Reviews. It is very important that you receive approval from required, applicable, institutional committees as well as be in contact with any ancillary departments that are included or impacted by your participation in the research project (e.g., Radiation Safety Committee, Institutional Biosafety Committee, Scientific Advisory Committee, Investigational Drug Services, Cancer Center Protocol Review and Monitoring Committee, etc.).
Research projects that utilize any UVMHN resources are required to have a coverage analysis and billing plan conducted by UVMHN Billing Compliance, so you should contact that office early in the process.
3. Meeting with UVM IRB Reliance Administrator
After completion of the local context review, the PI and his/her Designated Contact Person are required to meet with IRB staff such as the IRB Reliance Administrator. This meeting is to review:
• the Single IRB process
• your responsibilities as the UVM PI
• your responsibilities to the External IRB
• the appropriate consent/HIPAA document
• UVM expectations for continued submissions and communications
• the status and content of the reliance agreement
The meeting will include review of a “Study Team Meeting Regarding External IRB Reliance” checklist, and the UVM/UVMHN PI will be provided with a signed copy. Completion of this step does not mean that protocol activities may begin at UVM/UVMHN. Allowance to begin protocol activities at UVM/UVMHN will not occur until Step 6 in this process.
4. Submission of Local Materials for External IRB Review
The UVM Designated Contact is responsible for communicating with the Lead Site and/or External IRB to determine what documents are needed for review and the procedures for submission. It is expected that the required materials will have undergone local context review by the UVM IRB prior to this step although some Ancillary Reviews may be pending.
5. Once UVM PI Obtains External IRB Approval
Prior to the start of any research activities at UVM, the UVM PI must submit in UVMClick:
• External IRB approval memo for UVM/UVMHN site, PI, and local materials
• Final, External IRB-approved protocol (if different from version already submitted to UVM)
• Final, External IRB-approved local consent form(s)
• Any changes to local key personnel roster or the original Data Management and Security Plan since local review
The PI may not begin protocol activities until he/she receives notification from the UVM IRB that they have met the local requirements and are allowed to begin the protocol.
6. Allowance to Begin Research Activities Locally
The UVM IRB staff will review the final materials, as listed above, to ensure that all local requirements are met and that Ancillary Reviews are complete (i.e., UVMHN has completed a Coverage Analysis and Billing Plan if applicable, the final consent includes the required language, agreements are in place, etc.). Once it has been determined that all local issues are addressed, an “Approval to Begin Research Activities Reviewed by an External IRB” memo will be sent to the UVM PI, Primary Contact, and PI Proxies through UVMClick.
UVM IRB Ongoing Submission Requirements
a. Changes in PI or Key Personnel
UVM/UVMHN PI or Proxy must submit, in UVMClick, any changes to the local PI or key personnel. UVM is required to know who is assigned as the PI, as well as to ensure key personnel have completed required human subjects and good clinical practice (as applicable) training prior to working on the protocol.
b. Changes in Funding
UVM/UVMHN PI or Proxy must submit, in UVMClick, any changes to or new UVM funding for the project. The Sponsored Projects Administration Funding Proposal number (FP#) must be provided.
c. External IRB Determinations regarding Local Unanticipated Problems, Protocol Deviations, Noncompliance, Local Adverse Events, Participant Complaints, and Investigation by External IRBs or Federal Agencies (Reportable New Information reports)
See Section 18 of the UVM IRB Policies & Procedures for information regarding Reportable New Information requirements and timelines on External IRB studies at UVM. See Section 27 of the UVM IRB Policies & Procedures for information regarding Noncompliance procedures on External IRB studies at UVM.
d. Protocol changes that affect local required consent/HIPAA language
Protocol changes that affect UVM required consent/HIPAA language must be submitted to the UVM IRB for review prior to implementation. Submit the External IRB modification approval memo, the revised External IRB-approved protocol, and the revised local consent form for review. Note: Protocol changes that do not affect local required language do not require submission to UVM.
e. Closure of Protocol
UVM/UVMHN PI or Proxy must submit, in UVMClick, a notice of protocol or site closure.
Ongoing Monitoring
The UVM IRB will follow Section 26 of the UVM IRB Policies and Procedures for Human Subject Quality Assurance Reviews and the Noncompliance Policy and Procedures located in Section 27.
13.4 Non-Collaborative Review and UVM IRB
Revised 03/23/2023
The UVM IRB does not provide services to non-UVM or non-UVM Health Network entities or individuals when there is no collaborative relationship with researchers at UVM or UVMHN. We encourage outside entities to utilize an independent IRB as it is their sole purpose. There are many independent IRBs and they often have a variable fee dependent upon the level of risk for the project. UVM is familiar with the WCG IRB (formerly Western IRB or WIRB) and Advarra IRBs. Here is another website with information about independent IRBs: http://www.consortiumofirb.org/cirb_members.htm.
13.5 Procedures for Reviewing or Relying for NNE-CTR Grants (Sec __.114(b)(1))
The University of Vermont (UVM), MaineHealth (MH, including Maine Medical Center), University of Southern Maine (USM), and Northern New England Practice & Community Based Research Network Co-op (NNE PCBRN Co-op, including Dartmouth Hitchcock Medical Center) are collaborating organizations in the Northern New England Clinical and Translational Research Network (NNE-CTR). The NNE-CTR goals are to:
• Improve the infrastructure and support for clinical and translational research in Maine, New Hampshire, and Vermont
• Increase translational and clinical research capacity and efficacy by establishing collaborative and synergistic transdisciplinary research partnerships among the NNE-CTR institutions that emphasize health problems endemic in the rural populations of northern New England, including addiction, cancer, and cardiovascular disease, as well as the barriers that compromise rural healthcare delivery
• Establish innovative training, mentorship and professional development programs to enable clinical and translational scientists at the NNE-CTR institutions
• Develop infrastructure to support community healthcare practices and practice-based primary care research networks, particularly in rural areas, to engage clinical, community, public health and other stakeholders in the development and conduct of research relevant to the health needs of northern New England
Projects funded under this mechanism are required to use a single IRB. The collaborating institutions have agreed that for awarded Pilot Program projects, the lead institution for the pilot project will act as the Reviewing IRB. The collaborating Relying site(s) will cede their IRB review under the current SMART IRB Master Reliance Agreement.
Responsibilities
The Reviewing IRB will be responsible for regulatory review at the Expedited or Committee level ensuring that the protocol meets the regulatory requirements for protecting human subjects. The Relying sites are responsible for oversight of human subject protections at their respective institutions. Specific responsibilities are outlined in the SMART IRB Master Reliance Agreement.
Steps to Allow UVM IRB Reliance on an External IRB within the NNE-CTR
Guidance should be sought from the UVM IRB Reliance Administrator early in the process to ensure the request to rely is approvable and that all necessary steps are completed. The steps to allow reliance on an External IRB will be followed as described in Section 13.3 of the UVM IRB Policies and Procedures.
Steps for UVM to be the Reviewing IRB for other Sites on an NNE-CTR Grant
1. Request to become the Reviewing IRB
The protocol should be submitted to the UVM IRB through UVMClick following the standard submission process for a new study. See directions UVMClick User Guides, “Documentation for User Community,” and required UVM forms under IRB Forms Library “Initial Submission.” PIs should indicate on the Basic Information page in UVMClick that the UVM IRB will act as the Single IRB of record for other participating sites.
It must be clear in the protocol which sites are expected to rely on UVM’s IRB. The protocol should include information and procedures relevant to all sites. Other attachments should be relevant to the overall study (such as consent templates that can be used at all sites) or UVM/UVMHN-specific study conduct (such as local recruitment materials and Data Management and Security Plan). Documents specific to individual participating sites will be submitted later in the process.
As part of the submission, each site will have to submit their local context information relevant to the protocol. The form UVM IRB Required Local Context Form for Relying Sites should be completed by each site’s PI and IRB. This form will be provided to the UVM/UVMHN PI or Designated Contact by the UVM IRB Reliance Administrator early in the process to distribute to Relying sites, to allow ample time for completion.
2. Initial Study Review by UVM IRB and Ancillary Departments
The study will undergo a review by the UVM IRB to ensure it meets the regulatory requirements for protections of human subjects, in accordance with review procedures described in Section 3.1 and Section 3.2 of the UVM IRB Policies & Procedures. Ancillary Reviews will be conducted in accordance with Section 2 of the Policies & Procedures. Completion of initial study review does not mean that protocol activities may begin at participating sites. Each site must be added through a subsequent study modification after ceding reliance through the SMART IRB Online Reliance System.
3. Meeting with UVM IRB Reliance Administrator
After submitting protocol/consent materials to the UVM IRB for review and approval, the PI and his/her Designated Contact Person are required to meet with IRB staff. This meeting is to review the following items:
• the Single IRB process
• communication plans
• your responsibilities to the UVM IRB
• your responsibilities as the lead PI
• responsibilities of the relying institutions
• UVM expectations for continued submissions
Following the study team meeting, the UVM IRB will request that the other sites involved cede review to UVM IRB through the SMART IRB Online Reliance System.
4. Once UVM PI Obtains UVM IRB Approval for Overall Study
Once the UVM PI obtains initial study approval, the approved study documents must be uploaded by the PI into the SMART IRB Online Reliance System for review by the Relying Institutions. The Relying Institutions will review the reliance request and agree to cede review to the UVM IRB through the SMART IRB Online Reliance System.
Other methods for documentation of reliance using the SMART IRB Master Reliance Agreement will be considered on a case-by-case basis at the request of participating sites.
To request SMART IRB access, click on link below, select “Investigators: Request access” and follow directions on the subsequent screen. You will receive approval as a new user via email once approved.
The Designated Contacts and PIs at each participating site will need to create local documents (consent form/HIPAA authorization, recruitment materials, etc) for review and approval by UVM’s IRB. These documents are exchanged via email between sites. The UVM Designated Contact Person will review and then upload to Click for UVM IRB review and approval.
At this same time, data use agreements, as applicable, must be executed with the relying sites.
5. Relying Site Approval to Begin Activities
A formal approval, from the UVM IRB, for a relying site to begin local research activities will be sent to the UVM PI and Designated Contact Person through UVMClick. The relying sites may not begin protocol activities until they receive this formal approval notification from their contact at UVM and obtain all required approvals from their local IRB and institution. Additionally, the project should not begin until the PI has received confirmation of the NIGMS approval of the award from NNE-CTR Leadership. NIGMS confirms approval for funding after IRB approval is obtained for all sites participating in the research project.
UVM Ongoing Submission Requirements
UVM PIs should follow standard submission requirements for ongoing local IRB review and oversight of the study including protocol amendments and continuing reviews as applicable. Submissions must include information and documents from all relying sites. For instance, numbers of subjects accrued should include numbers from all sites broken down by site.
In addition to the standard local submissions, the UVM PI or Designated Contact Person must also submit Reportable New Information reports on behalf of relying sites as they will not have direct access to our system. The UVM PI must ensure that relying sites report Unanticipated Problems, protocol deviations, noncompliance, adverse events, participant complaints, and investigations by federal agencies in accordance with the UVM IRB’s reporting criteria (see Section 18 of the UVM IRB Policies and Procedures). Procedures found in Section 27 of the UVM IRB Policies and Procedures will be followed for all Noncompliance reviews.
Ongoing Monitoring
The UVM IRB will not conduct routine QA monitoring of Relying Sites. Relying sites are expected to have access to a QA program and are responsible for ongoing, local monitoring of the study. Dependent upon the complexity and risk level of the protocol, the UVM IRB may require something additional to the relying site’s plan for oversight.
Serious/Continuing Noncompliance Review Process
UVM IRB will conduct for-cause investigations as needed in accordance with Section 27 of the UVM IRB Policies and Procedures. Relying sites will be promptly notified if an audit or investigation will occur. Alternatively, the UVM IRB may request the Relying Sites to investigate issues of serious or continuing non-compliance and report the findings to UVM, or the UVM IRB and Relying Site may work cooperatively to conduct an audit/investigation. The Relying Sites will respond to all UVM IRB inquiries/clarifications and provide all requested records and information. The UVM IRB will inform the Relying Site of any corrective actions or required reporting as a result of the audit/investigation, and will provide reasonable opportunity for the Relying Site to review and comment on draft reports.
The UVM IRB is responsible for reporting to applicable regulators and sponsors.
13.6 Collaborative Research Between UVM and the VT Agency of Human Services (AHS)
Revised – 09/02/21
The VT Agency of Human Services and the University of Vermont (UVM) IRB have a reliance agreement in place that allows the AHS IRB to rely on the UVM IRB for collaborative projects led by a researcher at UVM
- Who has signed a scope of work with AHS; or
- Where AHS has provided financial support; or
- Where the project requests access to data or resources from one of the State member departments. Protocols that involve pregnant women, prisoners and children receiving services/support from AHS (e.g. research specifically targets women enrolled in the WIC program) must first be considered by the AHS IRB.
If you have a protocol that fits one of these scenarios, you should be in contact with the AHS IRB early in the process to determine if they wish to review the protocol or rely the UVM IRB review. The UVM IRB is required to review and approve regardless of the AHS IRB’s decision to review. However, the AHS IRB should always have the right to review prior to submission to the UVM IRB as any changes required by the AHS IRB must be included in the protocol materials submitted to the UVM IRB. In the situation where both IRBs review the protocol, you may not begin your activities until you have secured both the AHS and UVM IRB approvals. If the AHS IRB wishes to rely on the UVM IRB, researchers must provide confirmation from the AHS IRB that they will rely on the UVM’s review of the protocol under the current reliance agreement.
Communication between the UVM PI and the AHS IRB is imperative to ensure a smooth and timely review process through one or both IRBs.
Below is the link to the AHS IRB.
Example of protocol review flow is below.
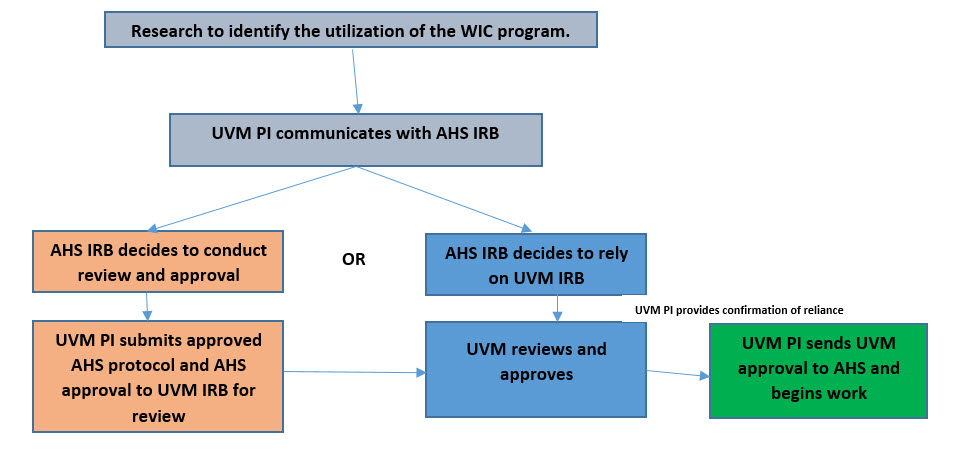
13.7 Collaborations with Community Partners
New 05.27.21
HHS regulations at 45 CFR 46.103(a) require that each institution engaged in HHS-conducted or -supported human subjects research provide written assurance, satisfactory to HHS, that it will comply with the requirements of the HHS regulations for the protection of human subjects, unless the research is exempt under 45 CFR 46.101(b). HHS regulations at 45 CFR 46.103(b) require that each institution engaged in HHS-conducted or -supported human subjects research certify to the HHS funding agency that the research has been approved by an IRB designated in the assurance.
A community partner is an individual employed at a community organization and/or an individual that is self-employed, in private practice or is otherwise involved where research is being conducted by a UVM PI. Typically, community partners are not affiliated with an academic institution and/or do not have a Federalwide Assurance (FWA) (here after referred to as a non-assured institution).
Under certain circumstances the UVM IRB may agree to extend their FWA to a non-assured institution.
Applicability
This mechanism applies to any Department of Health and Human Services conducted or sponsored human subjects research when the research is being conducted under the direction and supervision of the principal investigator from the assured institution, in this case UVM. The project must be
- led by a UVM PI;
- federally funded through UVM, and
- conducted within the State of Vermont.
This mechanism does not apply
- if the community partner intends to routinely conduct human subjects research; (FWA required)
- if the community partner is the prime awardee of a federal grant; (FWA required)
- if the project has a non-federal funding source; or
- if the community partner activities are determined to be not “engaged” in the research.
There are situations where a business can assist in the conduct of the study. The business may be engaged but the relationship is one of a contractual nature versus a collaborative nature. Portions of this policy may or may not apply dependent upon the situation. Contact the IRB office to discuss these types of scenarios.
Generally, the community partner would be considered “engaged” in human research, when for the purposes of nonexempt research, the community partner:
(1) obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or
(2) obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens; or
(3) obtains the informed consent of human subjects for the research.
Also, see Section 3.7 for other categories of engagement in research. If it is determined after consultation with an IRB Research Analyst that the community partner is engaged in research, UVM IRB may extend their FWA to cover their activity. If it is determined that they are not engaged in research, no further IRB review is necessary.
Types of Community Partners
Human subjects research conducted by UVM may involve the following two types of community partners:
1. A collaborating independent partner is:
- not otherwise an employee or agent of UVM/UVMMC;
- conducting collaborative research activities outside the facilities of UVM/UVMMC; and
- not acting as an employee of any institution with respect to his or her involvement in the research being conducted by UVM/UVMMC.
*payment, if applicable, will be made directly to the collaborating investigator
2. A collaborating institutional partner is:
- not otherwise an employee or agent of UVM/UVMMC;
- conducting collaborative research activities outside the facilities of UVM/UVMMC;
- acting as an employee or agent of a non-assured institution with respect to his or her involvement in the research being conducted by UVM/UVMMC; and
- employed by, or acting as an agent of, a non-assured institution that does not routinely conduct human subjects research.
*payment, if applicable, will be made to the collaborating institution
Conditions for Extending FWA to Collaborating Community Partners
Provision for covering individual collaborating partners from non-assured institutions under UVM’s FWA requires completion of the Individual Investigator Agreement (IIA). The IIA is signed by the collaborator and the UVM Institutional Official or delegate. An IIA covers only one collaborating partner. If multiple collaborating partners from non-assured institutions are participating in research involving human subjects, then separate IIAs must be executed for each collaborating partner. The UVM IRB will maintain the executed IIA(s) and provide to OHRP upon request. Please request the most current IIA template from your IRB Research Analyst.
Note: For collaborating institutional partners, the appropriate authorities at the non-assured institution also need to provide in writing that the conduct of the research is permitted at their institution.
The UVM PI responsibilities are outlined within the IIA and include, but not limited to:
- direction and supervision of all the collaborative research activities to be performed outside the assurance institution;
- applicable UVM and protocol training;
- making available to the collaborating partner regulatory documents, including the (a) The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research; (b) UVM’s FWA 00000723 found here and applicable Terms of the FWA; and (c) the relevant institutional policies and procedures for the protection of human subjects at UVM.
Educational Requirements for Approved Collaborating Partners
Individual collaborating partners must be listed as key personnel on the UVM protocol submission and complete the CITI Human Subjects Protection and Good Clinical Practice modules, and as applicable, hospital credentialing if the project requires access to any hospital information. To access the required UVM CITI training modules, collaborating partners will need to apply for a UVM NetID as an affiliate (instructions on the IRB website). The UVM IRB will consider the requirement for additional forms of education in the protection of human subjects on a case-by-case basis.
13.8 Procedures for Reviewing or Relying for NNE-CTR Grants
Revised 7/6/2023
(Sec __.114(b)(1)) The University of Vermont (UVM), MaineHealth (MH, including Maine Medical Center), University of Southern Maine (USM), and Northern New England Practice & Community Based Research Network Co-op (NNE PCBRN Co-op, including Dartmouth Hitchcock Medical Center) are collaborating organizations in the Northern New England Clinical and Translational Research Network (NNE-CTR). The NNE-CTR goals are to:
• Improve the infrastructure and support for clinical and translational research in Maine, New Hampshire, and Vermont
• Increase translational and clinical research capacity and efficacy by establishing collaborative and synergistic transdisciplinary research partnerships among the NNE-CTR institutions that emphasize health problems endemic in the rural populations of northern New England, including addiction, cancer, and cardiovascular disease, as well as the barriers that compromise rural healthcare delivery
• Establish innovative training, mentorship and professional development programs to enable clinical and translational scientists at the NNE-CTR institutions
• Develop infrastructure to support community healthcare practices and practice-based primary care research networks, particularly in rural areas, to engage clinical, community, public health and other stakeholders in the development and conduct of research relevant to the health needs of northern New England
Projects funded under this mechanism are required to use a single IRB. The collaborating institutions have agreed that for awarded Pilot Program projects, the lead institution for the pilot project will act as the Reviewing IRB. The collaborating Relying site(s) will cede their IRB review under the current SMART IRB Master Reliance Agreement.
Responsibilities
The Reviewing IRB will be responsible for regulatory review at the Expedited or Committee level ensuring that the protocol meets the regulatory requirements for protecting human subjects. The Relying sites are responsible for oversight of human subject protections at their respective institutions. Specific responsibilities are outlined in the SMART IRB Master Reliance Agreement.
Steps to Allow UVM IRB Reliance on an External IRB within the NNE-CTR
Guidance should be sought from the UVM IRB Reliance Administrator early in the process to ensure the request to rely is approvable and that all necessary steps are completed. The steps to allow reliance on an External IRB will be followed as described in Section 13.3 of the UVM IRB Policies and Procedures.
Steps for UVM to be the Reviewing IRB for other Sites on an NNE-CTR Grant
1. Request to become the Reviewing IRB
The protocol should be submitted to the UVM IRB through UVMClick following the standard submission process for a new study. See directions UVMClick User Guides, “Documentation for User Community,” and required UVM forms under IRB Forms Library “Initial Submission.” PIs should indicate on the Basic Information page in UVMClick that the UVM IRB will act as the Single IRB of record for other participating sites.
It must be clear in the protocol which sites are expected to rely on UVM’s IRB. The protocol should include information and procedures relevant to all sites. Other attachments should be relevant to the overall study (such as consent templates that can be used at all sites) or UVM/UVMHN-specific study conduct (such as local recruitment materials and Data Management and Security Plan). Documents specific to individual participating sites will be submitted later in the process.
As part of the submission, each site will have to submit their local context information relevant to the protocol. The form UVM IRB Required Local Context Form for Relying Sites should be completed by each site’s PI and IRB. This form will be provided to the UVM/UVMHN PI or Designated Contact by the UVM IRB Reliance Administrator early in the process to distribute to Relying sites, to allow ample time for completion.
2. Initial Study Review by UVM IRB and Ancillary Departments
The study will undergo a review by the UVM IRB to ensure it meets the regulatory requirements for protections of human subjects, in accordance with review procedures described in Section 3.1 and Section 3.2 of the UVM IRB Policies & Procedures. Ancillary Reviews will be conducted in accordance with Section 2 of the Policies & Procedures. Completion of initial study review does not mean that protocol activities may begin at participating sites. Each site must be added through a subsequent study modification after ceding reliance through the SMART IRB Online Reliance System.
3. Meeting with UVM IRB Reliance Administrator
After submitting protocol/consent materials to the UVM IRB for review and approval, the PI and his/her Designated Contact Person are required to meet with IRB staff. This meeting is to review the following items:
• the Single IRB process
• communication plans
• your responsibilities to the UVM IRB
• your responsibilities as the lead PI
• responsibilities of the relying institutions
• UVM expectations for continued submissions
Following the study team meeting, the UVM IRB will request that the other sites involved cede review to UVM IRB through the SMART IRB Online Reliance System.
4. Once UVM PI Obtains UVM IRB Approval for Overall Study
Once the UVM PI obtains initial study approval, the approved study documents must be uploaded by the PI into the SMART IRB Online Reliance System for review by the Relying Institutions. The Relying Institutions will review the reliance request and agree to cede review to the UVM IRB through the SMART IRB Online Reliance System.
Other methods for documentation of reliance using the SMART IRB Master Reliance Agreement will be considered on a case-by-case basis at the request of participating sites.
To request SMART IRB access, click on link below, select “Investigators: Request access” and follow directions on the subsequent screen. You will receive approval as a new user via email once approved.
https://reliance.smartirb.org/users/sign_in
The Designated Contacts and PIs at each participating site will need to create local documents (consent form/HIPAA authorization, recruitment materials, etc) for review and approval by UVM’s IRB. These documents are exchanged via email between sites. The UVM Designated Contact Person will review and then upload to Click for UVM IRB review and approval.
At this same time, data use agreements, as applicable, must be executed with the relying sites.
5. Relying Site Approval to Begin Activities
A formal approval, from the UVM IRB, for a relying site to begin local research activities will be sent to the UVM PI and Designated Contact Person through UVMClick. The relying sites may not begin protocol activities until they receive this formal approval notification from their contact at UVM and obtain all required approvals from their local IRB and institution. Additionally, the project should not begin until the PI has received confirmation of the NIGMS approval of the award from NNE-CTR Leadership. NIGMS confirms approval for funding after IRB approval is obtained for all sites participating in the research project.
UVM Ongoing Submission Requirements
UVM PIs should follow standard submission requirements for ongoing local IRB review and oversight of the study including protocol amendments and continuing reviews as applicable. Submissions must include information and documents from all relying sites. For instance, numbers of subjects accrued should include numbers from all sites broken down by site.
In addition to the standard local submissions, the UVM PI or Designated Contact Person must also submit Reportable New Information reports on behalf of relying sites as they will not have direct access to our system. The UVM PI must ensure that relying sites report Unanticipated Problems, protocol deviations, noncompliance, adverse events, participant complaints, and investigations by federal agencies in accordance with the UVM IRB’s reporting criteria (see Section 18 of the UVM IRB Policies and Procedures). Procedures found in Section 27 of the UVM IRB Policies and Procedures will be followed for all Noncompliance reviews.
Ongoing Monitoring
The UVM IRB will not conduct routine QA monitoring of Relying Sites. Relying sites are expected to have access to a QA program and are responsible for ongoing, local monitoring of the study. Dependent upon the complexity and risk level of the protocol, the UVM IRB may require something additional to the relying site’s plan for oversight.
Serious/Continuing Noncompliance Review Process
UVM IRB will conduct for-cause investigations as needed in accordance with Section 27 of the UVM IRB Policies and Procedures. Relying sites will be promptly notified if an audit or investigation will occur. Alternatively, the UVM IRB may request the Relying Sites to investigate issues of serious or continuing non-compliance and report the findings to UVM, or the UVM IRB and Relying Site may work cooperatively to conduct an audit/investigation. The Relying Sites will respond to all UVM IRB inquiries/clarifications and provide all requested records and information. The UVM IRB will inform the Relying Site of any corrective actions or required reporting as a result of the audit/investigation, and will provide reasonable opportunity for the Relying Site to review and comment on draft reports.
The UVM IRB is responsible for reporting to applicable regulators and sponsors.
14. Funding/Contracts/Fees
RPO, SPA and the Office of Clinical Trials Research work together to ensure all institutional and sponsor approvals and contracts are in place prior to the initiation of sponsored research involving human subjects.
The IRB is required to ensure that all research described in a grant application or proposal is entirely consistent with any corresponding protocol(s) reviewed and approved by the IRB. Any discrepancies must be resolved prior to the start of the project. The IRB works with SPA and the Office for Clinical Trials Research to establish that an appropriate connection is made between the application and the protocol being reviewed.
Relevant information regarding sponsored projects is shared between Offices (e.g. conflict of interest, study incentives, key personnel). Protocol approvals are not released until applicable contracts or agreements have been fully executed.
14.1 When the Project is a New Competing or a Competing Renewal Application and the New Protocol is Identical or Substantially Similar to an Approved Protocol
Obtaining grant funding is extremely competitive. The same grant proposal may be submitted to multiple funding agencies at once or the same agency at different time points. If you obtain new funding, it is your responsibility to submit the corresponding grant and protocol for IRB review and approval.
The IRB treats identical protocols as new applications, however, a new committee review may not be required if the project is the same or substantially similar to the previously approved protocol.
If this is the case, you must submit the following:
1. Complete the UVMClick eform and attach all applicable materials.
2. A letter to the committee chair explaining that you are submitting a similar grant application to a different funding agency. State that this new protocol application is identical to the old one (provide CHRBSS/CHRMS file #) with regard to hypotheses, specific aims, and human subject involvement (or describe minor differences).
If this application is essentially the same as the previously approved application with only minor differences clearly described in a letter, the protocol will receive administrative review. If substantial changes are proposed, then a new committee review may be required.
Competing Resubmissions or Supplements
Grant resubmissions require a modification to a previously approved protocol if it is identical or substantially similar to that protocol and grant. A copy of the resubmitted grant application can be submitted to the IRB with a modification request through UVMClick-IRB for review and approval.
Administrative and competitive supplements also require a modification to a previously approved protocol. A modification request can be submitted through UVMClick-IRB with a copy of the supplement for review and approval.
14.2 IRB Review of Just-in-Time (JIT) Protocols
What is a “Just-in-Time” (JIT) Request
The NIH just-in-time policy defers the submission of several proposal elements for grant submissions until after completion of Scientific Review and prior to Council Review and a decision of award. This policy is an effort to focus the NIH review on the science and to save the applicant time and effort submitting materials for a grant that might not be funded. One of the elements that can be withheld are protocol approvals.
A JIT notice is activated via an automated email from NIH to researchers with submissions that receive an impact score of 40 or less from the Scientific Review, regardless of the assigned Institute’s pay line. JIT requests are not a Notice of Award or even an indicator of possible funding. JIT requests are another step in the process of obtaining NIH funding.
Process for Obtaining Committee Approvals in the event of JIT Request
As soon as the researcher receives the automated notice from the NIH, he/she must evaluate whether or not the application has an impact score that merits committing time to the JIT process. NOTE: It is not necessary for the researcher to submit a protocol if the priority ranking is unfavorable.
If there is merit, the PI should contact their RPO analyst as soon as possible to discuss the recommended level of Committee review for the project. Full meetings are scheduled monthly. If the agenda allows, JIT protocols will be added to the next available convened meeting. The Committee does not have a mechanism to convene a separate meeting specifically for JIT. NIH, however, allows you to submit approvals at the earliest date they become available. You should be in communication with your program officer regarding timing of Committee approvals. While obtaining Committee approvals may delay an award it should not affect receipt of an award.
If the JIT request is a new or competing renewal and is identical or substantially similar to a previously approved protocol, see New Competing or Competing Renewal Grant Applications for further guidance.
If the JIT request is for a resubmission, see section New Competing or Competing Renewal Grant Applications for additional guidance.
14.3 Grant Proposals Lacking Definite Plans for Involvement of Human Subjects
Revised 1.30.2023
Certain types of applications may involve human subjects (within the funding period) but definite plans are not included in the application or protocol (45 CFR 46.118). This type of application would include such activities as institutional grants, training grants, and projects in which human subjects' involvement will depend upon completion of instruments, prior animal studies, or purification of compounds. Regulations give federal agencies and their grantee institutions the discretion to allow a limited release of federal research funding to investigators without approval or exempt status. Conditions include:
- Human subjects involvement will depend upon completion of significant pre-human subjects development activities, or
- The award is for a clinical research network or consortium that plans to add new protocols over the course of the award, or
- The award is for funds that will be awarded to specific projects that will be selected and funded by the awardee (e.g., a pilot project program; some training grants).
The IRB must certify that all human subjects research contained within a grant application has been reviewed and approved prior to release of funds, even if the plans for human subject involvement is unknown. The NIH refers to these as “delayed onset awards”. Therefore, investigators should submit the “Grant Proposals Lacking Definite Plans for Involvement of Human Subjects” form. “Delayed Onset of Research” review is recognized by the IRB only as a compilation of research being conducted under a specific grant. It does not constitute a review of the risk/benefit ratio of protocols to be conducted under it. Approval for these projects will be given a 6 month expiration date to ensure no human subject work has begun without a new IRB protocol submission. Once the new protocol has been submitted and given IRB approval the project may be closed. Funding can then be linked to the newly reviewed and approved protocol.
NIH guidance states that PIs are required to obtain prior approval from the sponsor for the addition of human subject research activity prior to implementation. See notice below for additional information. Those protocols (including informed consent documents) must be submitted for IRB review and approval separate from this request. The grant will also be reviewed and approved with the separate protocol submissions.
- Prior NIH Approval of Human Subjects Research in Active Awards Initially Submitted without Definitive Plans for Human Subjects Involvement (Delayed Onset Awards)
(NOT-OD-12-130) National Institutes of Health
No human subjects may be involved until the project has been reviewed and approved by the IRB and certification of approval submitted to the funding agency.
14.4 Contracts/Agreements
Protocols which are supported by an industry sponsor where money, materials (test articles, equipment, or other supplies), or intellectual property are exchanged require a contract or agreement be in place between the sponsor and either UVM or UVM Medical Center. Most industry-sponsored research contract review is done through the Office of Clinical Trial Research (OCTR) however a select few are handled through SPA.
The IRB requires a copy of the final contract prior to release of a protocol approval. OCTR/SPA will forward a copy of the final contract to the IRB who will review for potential conflicts of interest and appropriate subject payments. Many times this contract review is the final step to protocol approval and release, so researchers should plan accordingly and submit their contracts to the appropriate individuals early in the review process.
14.5 Changes to the Scope of a NIH Awarded Project
While most of the University’s NIH grants are under Expanded Authorities, eliminating the need for prior approval for most budget changes, NIH still requires prior approval before making changes that NIH considers changes in project scope.
The NIH Grant Policy Statement provides examples of actions requiring approval before they are made. Investigators should consult the complete NIH Grants policy statement for changes that may involve a change in scope.
In 2012 the NIH issued additional guidance for changes that involve human subjects in active awards and that will require prior NIH approval. (NOT-OD-12-129).
Some actions that may require prior approval as they represent a change in scope include:
- Change in the specific aims approved at the time of award
- Substitution of one animal model for another
- Any change from the approved use of animals or human subjects
- Shift of the research emphasis from one disease area to another
- Application of a new technology
- Transfer of the performance of substantive programmatic work to a third party if the third party is a foreign component
- Change in key personnel including withdraw from the project, absence during any continuous period of three-months or more, reduction in time devoted to the project by twenty-five percent or more.
Prior approvals may be requested by an email from a University Authorized Official to the project’s Grants Management Officer. If you would like to request prior approval for any of the changes mentioned above, please be in touch with your SPA administrator. Your administrator will advise you on the content of the email request, review it, and forward it to the University’s Authorized Official who will send it on to NIH.
14.6 Fees for Committee on Human Research Review of Sponsored Trials
Revised 11/18/20
Policy
The University’s Institutional Review Boards (IRBs) charge fees for initial review and annually for the life of the protocol for University of Vermont (UVM) and University of Vermont Medical Center (UVMMC) studies.
Fees will be applied to these types of protocols:
- Industry
- Pharmaceutical companies
- Other for profit entities
- Non-profit entities where such fees are not prohibited
- Investigator-initiated protocols with for profit sponsors
- Protocols initiated by affiliated Health Network sites
Fees will not be applied to these types of protocols:
- Federal or federal flow through
- State of Vermont
- Non-profit where fees are prohibited
- Investigator-initiated internally -funded studies.
IRB Fee Schedule
The fee schedule is reviewed annually and is subject to change.
Type of Fee | Description | 2009 Fee (effective 7/1/09) | 2019 Fee (effective 7/1/19) | 2020 Fee |
Initial Study Review | Initial study review by the convened IRB, or expedited member review (includes flat fee for all subsequent amendments) | $2,500 | $3,500 | No Change |
| Exempt Review | Exempt determination | $500 | ||
Administrative Annual Review | Annual review fees are incurred to cover all other follow-on submissions, RNIs, audits, compliance issues, submitted for the previous year (annual fees are not contingent upon whether there is a required continuing review) | $1,500 | $2,500 | No Change |
New Study, Reliance on External IRB | Subcommittee or administrative review (applicable when sponsors are for profit or not-for-profit that allow IRB fees) | NA | $1,000 | No Change |
Budgeting though the Office for Clinical Trials Research (OCTR) for IRB Fees
Contracts and budgets for industry or pharmaceutical-initiated projects are typically supported through OCTR. OCTR negotiates with sponsors to include the IRB fees in project budgets during the proposal negotiation processes. Many investigators like to include a “regulatory” fee as part of their budget. The regulatory fee covers the research coordinator cost of preparing materials for IRB review. This is a separate fee from the IRB fee and OCTR identifies these as separate line items in the budget.
Budgeting though Sponsored Projects Administration (SPA) for IRB Fees
Contracts and budgets for investigator-initiated with for profit sponsors and not for profit entities that do not prohibit IRB fees, are typically supported by SPA. There is a line item in the budget worksheet that addresses the required IRB fees. If the sponsor does not wish to provide fees, written justification must be provided. SPA will assist with negotiation of these fees. Many investigators like to include a “regulatory” fee as part of their budget. The regulatory fee covers the research coordinator cost of preparing materials for IRB review. This is a separate fee from the IRB fee should be identified as a separate line item in the budget.
Frequently Asked Questions
When will the initial fee be charged?
The initial IRB fee for both full and expedited review studies are invoiced once the initial protocol review is complete. Initial IRB approval will not be released to the researchers until the fee has been paid.
How are IRB fees paid?
For studies negotiated through OCTR, OCTR staff initiates payment through University of Vermont Accounting services.
For studies negotiated through SPA, IRB staff will charge the study account by electronic journal.
What happens if the contract or study is not approved?
The IRB fees are assessments of real costs associated with protocol review by the IRB. If subjects are never enrolled, the study terminates before milestones are met, expenditures exceed revenue, or a contract is never finalized, the investigator and department are responsible for all expenditures not covered by the sponsor, including the initial and any annual IRB fees.
When will the annual fee be charged?
The annual fees will be invoiced once per year after initial approval.
What if I wish to re-open a protocol after I closed it?
If you have requested that a protocol be re-opened after it has been closed, a new submission will be required and IRB fees will apply.
Will there be exceptions made to this policy?
Exceptions to this policy will be considered on a case-by-case basis by appropriate institutional officials. Requests for consideration must be submitted by the Principal Investigator or Sponsor in writing to the IRB. Requests for outright waiver of the fee must be received prior to protocol submission to the IRB.
15. Modification to Previously Approved Protocol
Requirement
Review of any changes to previously approved research is required by federal regulation [45 CFR 46.103(b)(4)] and is an essential element of the continuing review of research involving human subjects.
The IRB recognizes research is a continuous process and changes in the conduct of a study and/or changes to the consent document are necessary. However, no changes to an approved protocol should be implemented until the Committee has reviewed and approved the changes. This includes, but is not limited to, subject recruitment methods, consent form changes, treatment changes, modifications to the sponsor’s master protocol as well as changes or additions in study sites, investigators, or key personnel.
The CRC, PRMC and UVMCC also require review of changes to protocols under their purview. Check their respective websites for further guidance.
Major Modifications
Major modifications potentially affecting the risk/benefit ratio are reviewed through the full committee review process.
When modifications impact the safety of subjects previously enrolled who continue to receive study interventions, it may be necessary to convey this information (e.g., to obtain the consent of the subjects) by means of an addendum to the existing consent form or providing the subjects with an informational sheet regarding the update. The IRB may determine, for example, that such subjects must be notified of new findings or toxicities not noted at the time they were originally consented. Such notification is consistent with the view of informed consent as a continuous process and affords subjects the opportunity to determine whether or not they wish to continue their participation in the research. IRB policy is that a consent addendum be used instead of requesting the subject sign a full copy of the newly revised consent. Requesting subjects sign a full consent each time there is a revision is a practice that can confuse subjects unnecessarily. The IRB shall determine on a case-by-case basis when such notification, and its documentation, is required.
Major modification or a new protocol?
Sometimes the Committee must decide whether a new research activity should be considered as a major modification to an existing protocol or be developed as a stand-alone protocol. The committee will review the proposed changes with emphasis on the newly reviewed research activities
1. Purpose
2. population and
3. procedures
If only one is changing the submission could be viewed as an amendment but if all 3 are changing the Committee may recommend this activity be reviewed as a new protocol. The IRB will review all submissions on a case by case basis with CHRMS leadership and in consultation with the PI.
Minor/Administrative Modifications
Minor modifications not affecting the risk to subjects may be reviewed through the expedited review process.
Immediate Hazard
Federal regulations mandate that changes cannot occur until after IRB review and approval “except when necessary to eliminate apparent immediate hazards to the subject." The FDA has comparable criteria for implementing changes [21 CFR 56.108(a)(4)].
The definition of immediate hazard, as stated in the federal regulations for amendments, encompasses only those few instances where the immediate well-being of the subject is at risk. To that end, the subject’s well-being must benefit from
(1) continuing the research itself (rather than just discontinuing the research intervention and treating with standard of care) and
(2) the research must be changed immediately for the well-being of the subject.
Subjects may always be treated based on a physician’s determination of their needs but might not be eligible to continue in the research protocol. If a protocol is clinically faulted it should be corrected (through the amendment process) and the patient should be removed from the protocol and treated with the standard of care.
Example of “Immediate Hazard”: A subject has been enrolled on a local surgical protocol. The protocol requires that a certain immune-suppressing medication be administered to the subject during surgery, but the subject has an allergic reaction to that medication once surgery commences. The physician would determine the appropriate medical course of action and, if appropriate, the procedure would proceed.
Immediately following the procedure, the PI must:
- notify the IRB; then
- the PI must submit a Reportable New Information eform documenting the event;
- the PI should submit a Modification eform altering the protocol to allow for treatment or optional medications in case of allergic reactions (if appropriate); and
- the PI must ask the Committee to determine if the subject can be included in the study population as the protocol, as approved by the Committee, was not followed for this subject.
Requesting a Modification
Modification requests may be submitted at any time but must be approved before any changes can be implemented in the conduct of the protocol.
Submit a Modification eform to the Committee with all revised documents (i.e., protocol, questionnaires, recruitment flyers, consents, etc.) Please ensure you have included a revised date on your materials.
If the modification resulted in a revision to the consent form, the revised consent will be stamped with a new IRB approval date. This helps to track which consents apply to which version of the approved protocol.
Documentation
Once approved, the Committee will forward approval and a stamped consent form (if applicable) to the PI via email. Proof of the modification approval as well as the stamped consent form must be kept by the PI (perhaps in a Research Regulatory Binder) as evidence that the Committee has approved the change.
16. Continuing Review Requirements
Revised 4/6/2023
Federal Policy: Continuing review of research activity is required by federal regulations [(45 CFR 46.109 subpart (e)].
It is the policy of the IRB to review human research appropriate to the degree of risk involved, but not less than once per year. “Higher risk” research (as determined by the Committees on Human Research – hereafter “Committee”) may require more frequent reviews.
The purpose of continuing review is to determine:
1) whether the risks to subjects continue to be minimized and reasonable in relation to the anticipated benefits;
2) whether the selection of subjects continues to be equitable;
3) whether the informed consent continues to be appropriate;
4) changes in key personnel and whether mandatory training is complete;
5) whether there continue to be:
a. adequate provisions for monitoring the data collected to ensure the safety of the subjects, when appropriate;
b. adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data, when appropriate; and
c. appropriate safeguards for vulnerable populations.
Protocols that Do Not Require Continuing Review
Research protocols having a more than minimal risk determination (full committee review) are required to undergo a continuing review at least once a year. Continuing review is no longer required for some minimal risk research, including studies where the only remaining activity is the analysis of identifiable data/biospecimens or activity to obtain follow-up clinical data. Do not assume that you do not need to submit a continuing review, the IRB must make that determination.
While the majority of expedited studies will not require continuing review, there are a few exceptions where we will continue to require continuing review such as
• Any project regulated by the Food & Drug Administration (FDA)
• Any project where the sponsor requires continuing review
• Any protocol where safety findings justify additional oversight
• Grant Proposals Lacking Definite Plans for Involvement of Human Subjects has been received and requires tracking to ensure the PI subsequently submits the human subjects protocol as described in the grant application.
• The Committee is concerned with investigator compliance. In these instances, the investigator will be notified along with justification for the continuing review requirement.
• Protocols where the UVM IRB is the single IRB of record.
Ongoing Requirements When Continuing Review Is No Longer Required
While the 2018 change does not require continuing review, it does require that the investigator notify the IRB when the protocol is closed. The PI and study team must also continue to submit the following items in real-time:
• amendments for protocol changes
• adverse events, noncompliance and unanticipated problems
• DSMB and IDB reports
• key personnel roster updates
• protocol closure
• notice when personal identifiers are removed from the data/biospecimens and all codes and keys are destroyed
To assist researchers to remain in compliance with these ongoing submissions, the IRB has developed an Annual Protocol Review (DOCX) self-checklist located on our webpage The IRB will, additionally, remind researchers through our Newsletter, to review this checklist at least once annually.
When to Report
UVMClick-IRB will send a continuing review reminder when applicable to the PI approximately three months before the approval is due to expire. Reminders will be sent at two months and one month prior to expiration.
Researchers will be required to login into the system, complete the continuing review eform and submit.
Requirements during Continue Review
1. Upload or list protocol deviations not affecting risk to subjects that occurred during the review period. (examples here include visit off by one day, questionnaires not done)
Note: Examples of deviations that always require RNI reporting within 7 days include but are not limited to; unanticipated problem (UAP), local serious adverse reactions to drug or device, medication errors, breach of confidentiality, HIPAA deviations, consent deviations, research subject incarceration during study and research subject complaints.
2. Attach supporting documents: (Submit a copy of the last signed consent form of each type that was used (addendums) on this protocol over the last review period. Subject's name (only) should be blanked out leaving the date of consent for auditing purposes.
3. Attach all monitoring reports occurring in the last 12 months conducted by study sponsor or grant agency or lead institutional site.
As part of the continuing review process, the Committee may require that the research be restricted, modified, reviewed more frequently or terminated/suspended if risks have changed during the review period. Alternatively, special precautions or Committee-imposed restrictions, or shortened review periods, may be modified if current data support such actions. Ongoing approval will not be released until requested clarifications or changes have been received.
Documentation
Once approved, the Committee will return a signed Protection of Human Subjects Assurance to the PI/contact/proxy.
Continued Approval Policy
If there has been no activity on a research protocol after 3 years, the protocol will be withdrawn from the Committee’s consideration. Exceptions may be made if the funding period exceeds three years and the human subjects’ protocol is not scheduled to begin until after that time period. You must indicate that is the case on your continuing review form.
In addition, the committee will be administratively closing any non-treatment protocols in which there has been no activity reported within the last 5 years.
This policy does not apply to protocols that have been and plan to remain open to accrual but, to date, have not enrolled any subjects. (e.g. oncology group protocols that are approved for rare tumors). For that situation, the category “active - work in progress” should be checked on the continuing review form.
Expired Approvals
Extensions beyond the expiration date are not allowed by regulations.
a) If the Committee does not provide continued approval of the research by the specified expiration date, subject accrual is suspended pending continued approval of the research by the Committee. Note: A consent form should not be used to enroll new subjects until after the Committee reviews and approves a continuation of the research.
b) Where subjects are patients in treatment studies, continuation of research interventions or interactions is allowable for the safety and well-being of the individual and is reportable to the IRB.
c) Department chairs and Faculty Sponsors (if applicable) will be notified of a lapse in a researcher’s IRB protocol approval. The Committee views the lapse of protocol approval as noncompliance.
17. Closing a Protocol by the PI
Revised 5.17.2023
Investigators have the responsibility to formally close a study with current IRB approval once it is completed. This informs the IRB on the conduct and outcomes of the study, including any risks or problems that may have arisen since the last study renewal or approval and which may need to be disclosed to the study participants or the Committee.
Criteria for Closing a Protocol
Only close-out a study if all of the following conditions apply:
• Study is permanently closed to enrollment OR was never open for enrollment
• All subjects have completed all study-related interventions OR not applicable (e.g. study did not include interventions, no subjects were enrolled)
• Collection of private identifiable information is complete OR not applicable (no subjects were enrolled)
• Analysis of private identifiable information is complete OR not applicable (no subjects were enrolled)
Do not close a protocol if any of the following conditions apply:
• Actively enrolling and consenting
• Research-related interventions and/or follow-up is ongoing
• Biological specimens containing personally identifiable information or PHI are being maintained in a repository that has been approved as part of this study or upon which analysis or research is ongoing. If, however, specimens have been transferred to a separate repository that has ongoing IRB approval, the study may be closed.
• Primary data analysis or manuscript preparation that involves the use or access to personally identifiable information or PHI is ongoing.
• If the external study sponsor has not provided permission to close the study with the IRB.
Notification of Closure to the IRB
Notification must be done by completing a Request for Continuing Review eform in the UVMClick-IRB system. This provides the opportunity for the researcher to summarize all the activities into a final report. Researchers cannot use a modification eform to close a protocol as a final report is required.
Retention and Disposal of Data
In addition to informing the IRB of the closure, the PI must store the research records for the required length of time in accordance with the federal regulations, UVM policy, and any additional requirements stipulated by research sponsors and/or investigators’ professional associations. Data should be retained and disposed of according to the Data Management and Security Plan.
Subsequent Use of Data
Subsequent use of data collected under a closed protocol, whether by the original investigator or other investigators, may constitute human subjects research requiring IRB approval or a determination of Exemption from IRB review.
If the principal investigator is leaving the institution, it is the principal investigator’s responsibility to either close the study or transfer the protocol to another UVM/UVM Medical Center principal investigator. See Managing Research Prior to Departure information.
17.1 Closure of Protocol by Committee
The IRB may require that a protocol be closed in the following circumstances:
• If the work on a research protocol has not yet begun after a three-year period following IRB approval.
• Non-treatment protocols in which there has been no activity (no new enrollments and no current participants) within the last 5 years.
• If it is determined the principal investigator (including student researchers) is no longer affiliated with UVM/UVM Health Network. In this instance, the Faculty Sponsor and/or Department Chair will be contacted to close the protocol or to assign a new PI.
• Closure may be required because of noncompliance. This would only occur after IRB review and communication with the investigator. Termination of IRB approval is reportable to the appropriate federal department or agency head and institutional officials.
• A multisite protocol, where UVM relies on an External IRB, will be administratively closed upon notification from the Reviewing IRB indicating the protocol or the UVM site has been closed.
In any of the situations described above apply, the IRB office will notify the PI, as well as his/her department chair, of the study closure.
All protocol submission types (full, expedited, exempt, amendments) may be removed from committee consideration under the following circumstances:
• PI fails to respond to the IRB’s initial request for revisions and/or clarifications within 60 days while in UVMClick clarification requested (pre-review) state, prior to Committee or Designated review.
• PI fails to respond to the IRB’s request for revisions and/or clarifications within 60 days while in the modifications required state, after Committee or Designated review.
• Ancillary reviewer(s) fail to respond to the IRB’s request for review within 10 months while in post review.
External Protocols (Single IRB)
• A new multisite protocol, where UVM relies on an External IRB, will be withdrawn from consideration if the UVM PI fails to respond to the UVM IRB’s initial requests for local context review revisions and/or clarifications within 60 days while in UVMClick clarification requested (pre-review) state, prior to Facilitated Review or recording the sIRB decision.
• A new multisite protocol, where UVM relies on an External IRB, will be withdrawn from consideration if the UVM PI fails to respond to the UVM IRB’s requests for local context review revisions and/or clarifications within 60 days while in UVMClick modifications required (pending sIRB review or post-review) state, after Facilitated Review or sIRB review.
• New multisite protocols, where UVM relies on an External IRB, will be withdrawn from consideration if the sIRB and/or ancillary reviewer(s) fail to respond to the UVM IRB or UVM Study Team’s request for review within 10 months while in pending sIRB review or post-review state.
• New multisite protocols, where UVM relies on an External IRB, will be withdrawn from initial consideration upon notification from the Reviewing IRB or Lead Study Team indicating the protocol has been closed.
Notification of Committee Closure or Removal of Protocol
In any of the situations described above, the IRB office will notify the PI, as well as his/her department chair, prior to study withdrawal or closure.
Contact your IRB Regulatory Analyst or IRB Reliance Administrator if sponsors, individuals, or processes outside of your control do not allow adherence to the response timeline criteria. Exceptions to response timeline criteria may be allowed on a case-by-case basis.
What If I Need to Reopen a Closed Protocol?
If an investigator needs to reopen a protocol after it has been formally closed with the IRB, the investigator would be required to submit a new protocol for review and approval. If the study is billable, the IRB will invoice for this new review.
18. Reportable New Information
Revised 09/11/23
Requirement
Review of reportable new information (RNIs) involving risk to subjects or others is required by federal regulations (45 CFR 46 and for FDA regulated articles 21 CFR 312 and 21 CFR 812), and is an essential element in the protection of research participants.
Reportable New Information
Reportable New information is information that may impact the conduct of an IRB-approved protocol, or the safety and welfare of the participants enrolled in that protocol. Below are examples of reportable new information that must be submitted to the IRB for review and determination.
1) Unanticipated Problems Involving Risks to Subjects (including death or an adverse event that meet all three of the following criteria)
1. Is unexpected (in terms of nature, severity, or frequency) given (a) the procedures described in the research protocol documents (e.g., the IRB-approved research protocol and informed consent document) and (b) the characteristics of the human subject population being studied.
2. Is related or possibly related to participation in the research (in this Instruction, possibly related means there is a reasonable possibility that the incident, experience, or outcome may have been caused by the procedures involved in the research).
3. Suggests that the research places human subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized.
2) Interim findings -- These include reports, action letters, report of interim results, device malfunction, or any other finding that indicates an unexpected adverse change to the risks or potential benefits of the research.
3) Protocol deviations when the safety and/or wellbeing of the participant is potentially compromised.
4) Complaint -- A research-related complaint by a research subject or another person
5) Study Personnel -- omissions of personnel from the approved protocol when they are conducting research
6) Audit inspection, or inquiry by a federal agency (e.g., FDA Form 483).
7) Breach of Confidentiality - Resulting from disclosure of confidential information or identifiable private information or lost/stolen confidential information (lost laptop, inadvertent email distribution)
8) Incarceration of a subject in a study not approved by the IRB to involve prisoners.
9) Suspension or premature termination by the sponsor, investigator, institution, or other IRB.
10) Quality Assurance Reports
11) High Risk Protocols - The IRB may, in coordination with other institutional oversight committees, categorize a protocol as “higher risk” and require the Investigator to follow a specific “high risk” reporting procedure. This high-risk determination will be made at time of initial review or any time after initial review if the IRB feels it is warranted. This determination and the requirements will be clearly communicated back to the Investigator. Examples of higher risk protocols of clinical trials for which the IRB may institute more stringent reporting are local, investigator-initiated early phase (Phase I, Phase I/II) study without a DSMB; local, investigator-initiated trial in extremely vulnerable populations, e.g., very sick patients, subjects unable to consent for themselves, prisoners. If a protocol is required to follow a high-risk reporting procedure, those reports must be submitted to the IRB utilizing the Reportable New Information eform.
12) Improper consent process or wrong form -- always has the potential to increase risk to subjects
13) Intentional change to the protocol without IRB approval to eliminate apparent immediate hazard to subject
14) Medication or Laboratory Errors administered as part of the research that involved increased risk to subjects
Do not submit to the IRB
1. IND SAFETY REPORTS AND STUDY PROGRESS REPORTS
Individual IND Safety Reports do not necessarily meet the reportable criteria and are recognized by OHRP and the FDA to not yield information that is useful to IRBs. The reports often lack context and detail, are often incomplete and unanalyzed, and as such, inhibit an IRB’s ability to assure the protection of human subjects. Therefore, IND safety reports do not require submission to the IRB. Similarly, study progress reports do not provide any additional safety information and are not reportable to the IRB.
However, if an individual IND report results in a revision to the protocol or consent, a Reportable New Information eform must be submitted to the IRB with the specific IND safety report. A separate modification request to make the required changes along with applicable materials must also be submitted.
2. RNIs that occur at other institutions who are conducting the same protocol do not require submission to our IRB unless UVM is the IRB of record. RNIs occurring under a multicenter study should be reviewed in accordance with a monitoring plan described in the lead institution’s IRB-approved protocol.
3.Good Clinical Practice errors
While investigators in LCOM and researchers conducting clinical trials need to adhere to good clinical practice (GCP), it is not required that GCP errors be reported as an RNI.
Notification Timelines for Reporting
RNIs are to be reported within 7 business days of discovery. If needed information is not available within 7 days discovering the issue, submit an initial report (indicating it is an initial report) and then follow-up with the IRB as soon as information becomes available.
IRB Submission, Review and Communications
Local RNIs meeting any of the examples above must be submitted through the UVMClick system as an RNI. The report should include an explanation of the situation, the actual or potential for harm, the findings of any investigation, and corrective actions. Any requests for clarification and/or responses must be sent through the system.
RNIs will be initially reviewed by a IRB Regulatory Analyst to determine how the report is to be processed. RNIs that potentially meet the criteria as serious or continuing noncompliance will be reviewed following sections 27.
Noncompliance Policy and 27.1 Noncompliance Review Procedures. All other RNI reviews will follow procedures in section 1.3.1 Safety Subcommittee Review.
Documentation
The PI must keep a copy of the Reportable New Information as evidence of IRB submission.
18.1 Reportable New Information on External IRB Studies
When relying on an External IRB (Reviewing IRB, IRB of Record, Single IRB) it is important to always follow that IRB’s reporting requirements for submission of reportable new information, in addition to the UVM guidance below.
The External IRB’s requirements may be different from the UVM’s requirements, so UVM/UVMHN PIs and Designated Contact Persons should ensure they have access to the External IRB’s policies and procedures early in the reliance process. Reporting expectations might also be addressed in reliance agreements.
Requirements and Timelines for Reporting to the UVM IRB When Relying on an External IRB
UVM/UVMHN researchers should reference the following table to determine if a local event requires reporting to the UVM IRB.
If a researcher has questions about whether a specific local event must be reported, they should contact the UVM IRB Reliance Administrator.
Reportable new information submissions should include all available, relevant documentation (i.e. determination memo, approved CAPA) from the External IRB.
See Section 18.0 and Section 27.2 of the Policies and Procedures for definitions and examples of the following events.
| Event | Report to the UVM IRB? | Timeline for Reporting to the UVM IRB |
|---|---|---|
| Determination by the External IRB of: Unanticipated Problem (UAP) involving risk to subjects or others | Yes | Within 7 business days of receiving the External IRB’s determination |
| Determination by the External IRB of: Serious or Continuing Noncompliance | Yes | Within 7 business days of receiving the External IRB’s determination |
| Notification by Sponsor, Lead PI/Site, or External IRB of: Suspension or premature termination | Yes | Within 7 business days of receiving the notification |
| Review by the External IRB of: Research-related complaint by a research participant or another person | Yes | Within 7 business days of receiving the External IRB’s determination or acknowledgement |
| Audit, inspection, or inquiry by the External IRB for possible serious or continuing noncompliance | Yes | Within 5 business days of learning about the inspection |
| Audit, inspection, or inquiry by a federal agency and any written reports from federal agencies (e.g., FDA form 483) | Yes | Within 5 business days of learning about the inspection or receiving the report |
| Breach of Confidentiality (including breaches involving Protected Health Information regardless of whether UVM is the Privacy Board) | Yes | Within 7 business days of discovering the event |
| Interim findings | No | n/a |
| Protocol, consent, or eligibility deviations prior to External IRB Review and Determination | No | n/a |
| Omission of personnel from the approved protocol, when they are conducting research | No | n/a |
| Incarceration of a subject in a study not approved by the IRB to involve prisoners | No | n/a |
| Reports resulting from routine Quality Assurance audits conducted by the External IRB or Sponsor (unless the External IRB makes a final determination of UAP or Serious or Continuing Noncompliance) | No | n/a |
19. Incidental Findings in Neuroimaging Protocols – Detection and Management
UVM has determined that MRI procedures that involve the administration of intravenous contrast, sedation, or drugs are greater than minimal risk since the probability and magnitude of harm or discomfort anticipated in the research are greater than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests [45 CFR 46.102(i)]. These protocols require full review.
For studies involving MRI scans without intravenous contrast, sedative or drug administration in healthy subjects, or in a disease/condition when the scan does not pose any additional risk, the study may be found to present no greater than minimal risk. This means the probability and magnitude of harm or discomfort anticipated in the research are not greater than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. These protocols can undergo expedited review.
This document reflects standard procedures accepted by the IRB for MRI research imaging. It provides guidelines for how to describe the procedures in the protocol and consent form. These standards do not apply to therapeutic imaging.
MRI Risk Information for Researchers
Risks due to the static magnetic field of the scanner:
The powerful magnetic field of the scanner can attract certain metallic objects known as “ferromagnetic” objects, causing them to move suddenly and with great force towards the center of the magnet. This may pose a risk to the patient or anyone in the way of the object, and has resulted in several deaths worldwide. Care must be taken to prevent ferromagnetic objects from entering the MR scan room. It is vital that anyone entering the scan room removes metallic objects, including watches, jewelry, and items of clothing that have metallic threads or fasteners.
The powerful magnetic field of the scanner can also exert a force on metallic implants and other materials inside the subject’s body, including (but not limited to) aneurysm clips, metallic fragments in the eye, and bullets. Movement of such objects can cause serious injury or death. Researchers must screen potential subjects for possible metal in their body and assume that such metal is unsafe, unless it has been approved by qualified personnel.
Risks due to time-varying magnetic field gradients:
MRI uses electrical currents to generate magnetic gradients used to acquire images. This results in the loud sounds associated with MRI. These rapidly varying magnetic fields can also induce electrical currents into conducting wires, such as cardiac monitoring leads and implanted electrical devices (which might cause the device to fail). For safety, subjects are required to wear earplugs or headphones to protect their hearing; patients with implantable electrical devices are not to be scanned; and care must be taken to use only MRI-compatible electrical devices. Correct placement of leads and devices is essential to their safe operation.
Risks due to radio-frequency (RF) power:
The MRI uses RF transmission and reception at similar frequencies to those used for FM radio. Some of this RF energy is absorbed by the body, and may cause a small temperature rise. Additionally it can cause electrically conducting materials such as aluminum foil to heat up, and has caused severe burns to patients. The scanner is designed to operate at FDA-approved limits on patient heating. RF can also cause electrically conducting materials such as aluminum foil to heat up, and have caused severe burns to patients. Therefore, medication patches that may contain aluminum backing should not be worn during the MRI scan; metallic tattoos must be evaluated for safety; and extra caution should be exercised in patients with poor temperature regulation. Research subjects should be informed that this may be a risk and that they can ask for increased fan speed or to use a light sheet/blanket during the scan
Considerations for Special Populations:
Pregnancy: There is no known risk of MRI for pregnant women or to the developing fetus, and no known mechanism of potential risk under normal operating procedures with a magnet of 4 Tesla or less. Nonetheless, the possibility that risks may be discovered in the future cannot be ruled out. There is increased risk for pregnant women/fetuses of contrast-enhanced scans, and pregnancy must be an exclusion for contrast-enhanced studies unless there is direct benefit. Protocols with non-contrast enhanced MRI scans must include that there is no known risk to pregnant women and fetuses, but that risks may be discovered in the future. Protocols with non-contrast enhanced MRI scans must provide justification(s) for either inclusion of, or theexclusion of pregnant women. . If pregnant women are to be excluded, the protocol must describe the mechanism for screening for pregnancy. Protocols with contrast-enhance MRI scans must exclude pregnant women if there is no direct benefit from the study.
Minors: The concerns for minors include proper consent/assent; a plan for sharing sensitive information with parents (if applicable); and recommendations regarding obtaining medical history information from children.
Risks due to the use of MRI contrast agents
FDA-approved gadolinium-based contrast agents
Gadolinium contrast agents have been approved for use since the late 1980s. Although these agents can be differentiated on the basis of stability, viscosity, and osmolality, they cannot be differentiated on the basis of efficacy. Gadolinium chelates are extremely well tolerated by the vast majority of patients in whom they are injected. Acute adverse reactions are encountered with a lower frequency than is observed after administration of iodinated contrast media.
When gadolinium-based contrast is being used, subjects need to be screened for possible kidney or liver impairments and excluded appropriately.
Patients who answer yes to the following criteria must have had a blood serum creatinine drawn within 60 days of the MRI scan to determine if gadolinium can be safely administered:
- Renal disease history (including solitary kidney, renal transplant, renal tumor)
- Liver disease
- Age >60
- History of hypertension
- History of diabetes
It is the Principal Investigator’s responsibility to obtain the above laboratory values prior to subject’s research scan.
Gadolinium contrast administered to patients with acute renal failure or severe chronic kidney disease can result in a syndrome of nephrogenic systemic fibrosis (NSF).
Nephrogenic systemic fibrosis (NSF) is a fibrosing disease, primarily involving the skin and subcutaneous tissues but also known to involve other organs, such as the lungs, esophagus, heart, and skeletal muscles. Initial symptoms typically include skin thickening and/or pruritis.
Deposits of gadolinium can accumulate in the brain, skin and bone. Deposits of gadolinium remain in the brains of some patients who have undergone 4 or more MRI scans for a prolonged time after the last administration. It is unknown whether these deposits are harmful or can lead to adverse health effects.
Other contrast agents
If your research study involves the use of an investigational agent or an agent other than gadolinium, the risk section must be specific to the agent being studied. Please check with your sponsor or the package insert. Consult with MRI staff if you are unsure about the contrast needs for your research.
Common Contraindications to MRI
The magnetic field of the MRI environment has the potential to cause burns or bodily injury if ferrous metal objects are implanted in the body or if personal articles containing ferrous material are brought into the environment. The protocol must specify how participants with contraindications for MRI studies (internal/implanted defibrillator or pacemaker, surgical brain aneurysm clips, cochlear implant, or known metal fragments embedded in the body) will be excluded.
The protocol must specify how participants with medical or electronic devices that may interfere with the scan or pose a risk will be evaluated and how risks to these participants will be minimized. Examples of such devices include but are not limited to:
- Artificial heart valves;
- Implanted drug infusion ports;
- Artificial limbs or metallic joint prostheses;
- Implanted nerve stimulators;
- Metal pins, screws, plates, stents or surgical staples.
Incidental Findings
There is a risk that the image will reveal a potentially clinically important finding, but which is beyond the aims of the study. In the event of the confirmation of a significant anomaly, this information will likely be distressing to the subject and constitutes a psychological risk. Additionally, see “Incidental Findings in Neuroimaging Protocols – Detection and Management”
University and UVM Medical Center researchers must make adequate provisions for monitoring the data collected to ensure the safety of subjects. In the course of study monitoring, information incidental to the research goals may be identified which may impact the safety and/or wellbeing of the subjects. Scientists and clinicians should anticipate the potential for incidental findings in experimental design and establish a process to handle the discovery of an incidental finding.
Incidental Finding – Definition
A finding discovered in the course of research participation for which there is potential health importance. An incidental finding is beyond the specific aims of the protocol.
Note: The UVM MRI Research Center 3T magnet is not considered a clinical magnet and therefore neither the images nor reports can be incorporated into the subjects’ clinical medical record and should not be used for diagnosing or treating medical conditions. Any abnormality found utilizing this magnet would be considered an incidental finding.
IRB Requirements Regarding Protocols Which May Have Incidental Findings
The IRB requires that the way in which incidental findings will be handled is made explicit in the study design. The IRB also requires that the way in which incidental findings will be handled is made explicit to any potential research subject through the informed consent process.
Research x-rays that are included in a protocol should not be designed nor intended to detect health problems in subjects. The x-rays required as part of a research study do not substitute for an appropriate medical examination by a qualified health care provider. The information from these x-rays should not be shared with the subject or their personal physician, unless there is an incidental finding.
Disclosure of clinically meaningful findings should be conducted by a licensed physician (or psychologist, genetic counselor, or other professional as appropriate) whenever possible. The person responsible for communicating an incidental finding must be referenced in the plan for disclosure submitted to the IRB.
Consent Template Section
Incidental Findings
There is a possibility that while reviewing your (insert test) we may see an abnormality that may have health implications that we did not expect to see. This is what is called an “incidental finding.”
If we see an incidental finding, a qualified person (usually a member of the research team) will communicate the information to you. If you wish, we will provide information about this incidental finding to your primary doctor or we will refer you to an appropriate doctor for further evaluation.
This study is neither designed nor intended to detect health problems. The imaging that you will have as part of this research study does not substitute for an appropriate medical examination by a qualified health care provider. If you suspect that you might be suffering from injury or illness, you should not rely on this study as a way to determine your health status. The information from this image will not be shared with you or your personal physician, unless (as mentioned above) there is an incidental finding.
An incidental finding may cause you to feel anxious. If you have further tests done, those results will then become part of your medical record, which may affect current and future health or life insurance. The costs for any care that will be needed to diagnose or treat an incidental finding would not be paid for by this research study. These costs would be your responsibility.
19.1 Standards and Language for Studies Involving MRI
UVM has determined that MRI procedures that involve the administration of intravenous contrast, sedation, or drugs are greater than minimal risk since the probability and magnitude of harm or discomfort anticipated in the research are greater than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests [45 CFR 46.102(i)]. These protocols require full review.
For studies involving MRI scans without intravenous contrast, sedative or drug administration in healthy subjects, or in a disease/condition when the scan does not pose any additional risk, the study may be found to present no greater than minimal risk. This means the probability and magnitude of harm or discomfort anticipated in the research are not greater than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. These protocols can undergo expedited review.
This document reflects standard procedures accepted by the IRB for MRI research imaging. It provides guidelines for how to describe the procedures in the protocol and consent form. These standards do not apply to therapeutic imaging.
MRI Risk Information for Researchers
Risks due to the static magnetic field of the scanner:
The powerful magnetic field of the scanner can attract certain metallic objects known as “ferromagnetic” objects, causing them to move suddenly and with great force towards the center of the magnet. This may pose a risk to the patient or anyone in the way of the object, and has resulted in several deaths worldwide. Care must be taken to prevent ferromagnetic objects from entering the MR scan room. It is vital that anyone entering the scan room removes metallic objects, including watches, jewelry, and items of clothing that have metallic threads or fasteners.
The powerful magnetic field of the scanner can also exert a force on metallic implants and other materials inside the subject’s body, including (but not limited to) aneurysm clips, metallic fragments in the eye, and bullets. Movement of such objects can cause serious injury or death. Researchers must screen potential subjects for possible metal in their body and assume that such metal is unsafe, unless it has been approved by qualified personnel.
Risks due to time-varying magnetic field gradients:
MRI uses electrical currents to generate magnetic gradients used to acquire images. This results in the loud sounds associated with MRI. These rapidly varying magnetic fields can also induce electrical currents into conducting wires, such as cardiac monitoring leads and implanted electrical devices (which might cause the device to fail). For safety, subjects are required to wear earplugs or headphones to protect their hearing; patients with implantable electrical devices are not to be scanned; and care must be taken to use only MRI-compatible electrical devices. Correct placement of leads and devices is essential to their safe operation.
Risks due to radio-frequency (RF) power:
The MRI uses RF transmission and reception at similar frequencies to those used for FM radio. Some of this RF energy is absorbed by the body, and may cause a small temperature rise. Additionally it can cause electrically conducting materials such as aluminum foil to heat up, and has caused severe burns to patients. The scanner is designed to operate at FDA-approved limits on patient heating. RF can also cause electrically conducting materials such as aluminum foil to heat up, and have caused severe burns to patients. Therefore, medication patches that may contain aluminum backing should not be worn during the MRI scan; metallic tattoos must be evaluated for safety; and extra caution should be exercised in patients with poor temperature regulation. Research subjects should be informed that this may be a risk and that they can ask for increased fan speed or to use a light sheet/blanket during the scan
Considerations for Special Populations:
Pregnancy: There is no known risk of MRI for pregnant women or to the developing fetus, and no known mechanism of potential risk under normal operating procedures with a magnet of 4 Tesla or less. Nonetheless, the possibility that risks may be discovered in the future cannot be ruled out. There is increased risk for pregnant women/fetuses of contrast-enhanced scans, and pregnancy must be an exclusion for contrast-enhanced studies unless there is direct benefit. Protocols with non-contrast enhanced MRI scans must include that there is no known risk to pregnant women and fetuses, but that risks may be discovered in the future. Protocols with non-contrast enhanced MRI scans must provide justification(s) for either inclusion of, or theexclusion of pregnant women. . If pregnant women are to be excluded, the protocol must describe the mechanism for screening for pregnancy. Protocols with contrast-enhance MRI scans must exclude pregnant women if there is no direct benefit from the study.
Minors: The concerns for minors include proper consent/assent; a plan for sharing sensitive information with parents (if applicable); and recommendations regarding obtaining medical history information from children. Please see sections 9.1 of this research manual for more information.
Risks due to the use of MRI contrast agents
FDA-approved gadolinium-based contrast agents
Gadolinium contrast agents have been approved for use since the late 1980s. Although these agents can be differentiated on the basis of stability, viscosity, and osmolality, they cannot be differentiated on the basis of efficacy. Gadolinium chelates are extremely well tolerated by the vast majority of patients in whom they are injected. Acute adverse reactions are encountered with a lower frequency than is observed after administration of iodinated contrast media.
When gadolinium-based contrast is being used, subjects need to be screened for possible kidney or liver impairments and excluded appropriately.
Patients who answer yes to the following criteria must have had a blood serum creatinine drawn within 60 days of the MRI scan to determine if gadolinium can be safely administered:
- Renal disease history (including solitary kidney, renal transplant, renal tumor)
- Liver disease
- Age >60
- History of hypertension
- History of diabetes
It is the Principal Investigator’s responsibility to obtain the above laboratory values prior to subject’s research scan.
Gadolinium contrast administered to patients with acute renal failure or severe chronic kidney disease can result in a syndrome of nephrogenic systemic fibrosis (NSF).
Nephrogenic systemic fibrosis (NSF) is a fibrosing disease, primarily involving the skin and subcutaneous tissues but also known to involve other organs, such as the lungs, esophagus, heart, and skeletal muscles. Initial symptoms typically include skin thickening and/or pruritis.
Deposits of gadolinium can accumulate in the brain, skin and bone. Deposits of gadolinium remain in the brains of some patients who have undergone 4 or more MRI scans for a prolonged time after the last administration. It is unknown whether these deposits are harmful or can lead to adverse health effects.
Other contrast agents
If your research study involves the use of an investigational agent or an agent other than gadolinium, the risk section must be specific to the agent being studied. Please check with your sponsor or the package insert. Consult with MRI staff if you are unsure about the contrast needs for your research.
Common Contraindications to MRI
The magnetic field of the MRI environment has the potential to cause burns or bodily injury if ferrous metal objects are implanted in the body or if personal articles containing ferrous material are brought into the environment. The protocol must specify how participants with contraindications for MRI studies (internal/implanted defibrillator or pacemaker, surgical brain aneurysm clips, cochlear implant, or known metal fragments embedded in the body) will be excluded.
The protocol must specify how participants with medical or electronic devices that may interfere with the scan or pose a risk will be evaluated and how risks to these participants will be minimized. Examples of such devices include but are not limited to:
- Artificial heart valves;
- Implanted drug infusion ports;
- Artificial limbs or metallic joint prostheses;
- Implanted nerve stimulators;
- Metal pins, screws, plates, stents or surgical staples.
Incidental Findings
There is a risk that the image will reveal a potentially clinically important finding, but which is beyond the aims of the study. In the event of the confirmation of a significant anomaly, this information will likely be distressing to the subject and constitutes a psychological risk. Additionally, see “Incidental Findings in Neuroimaging Protocols – Detection and Management”
20. Investigational Drugs (including Biologics)
Revised 10/02/20
The United States Food and Drug Administration’s Investigational New Drug (IND) program is the means by which an entity or investigator obtains permission to administer an investigational drug to human subjects (or an approved drug used for a new indication or new population of patients) (21 CFR 312). The FDA has two primary objectives in reviewing an IND: 1) to assure the safety and rights of subjects in all phases of an investigation and 2) in phases 2 and 3, to help assure that the quality of the scientific evaluation of the drug is adequate to permit an evaluation of the drug’s effectiveness and safety.
All studies that use a drug not approved for marketing by the FDA will always require an IND. By a rather broad set of definitions for a “new drug,” all studies using not only new molecular entities or unapproved pharmaceuticals but also approved drugs used in unapproved indications, in new formulations, in new dosages, in a patient population that would be put at increased risk require an IND. Under specific criteria, an exemption from the IND requirement may be met (discussed later).
INDs may be required for research introducing food or food-derived products, spices/herbs or dietary supplements. See section 20.3 for additional information.
Types of INDs
Commercial INDs are filed by companies to obtain marketing approval for a new drug.
Research or Investigator INDs are non-commercial INDs filed by researchers to study an unapproved drug or to study an approved drug for a new indication or new patient population.
Expanded Access INDs
Emergency Use INDs (compassionate use or single-patient) are filed for emergency use of an unapproved drug when the clinical situation does not allow sufficient time to submit an IND in accordance with the regulation. These are most commonly used for life-threatening conditions for which there is no standard treatment.
Intermediate-size Population INDs are filed for use by more than one patient, but generally fewer patients than are treated under a typical treatment IND or protocol. The investigational product may or may not be under development for marketing.
Treatment INDs are filed to make drug available for a large population of patients with serious or immediately life-threatening conditions. The investigational product must be under active development for marketing.
Screening INDs are filed for multiple, closely related compounds in order to screen for the preferred compounds or formulations. The preferred compound can then be developed under a separate IND.
Definitions
Drug – The Food, Drug, and Cosmetic Act (FD&C Act) defines drugs as “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease” and as “articles (other than food) intended to affect the structure or any function of the body of man or other animals.
Food – The FD&C Act defines food as “articles used for food and drink for man or animals, chewing gum, and articles used for components of any such article.
Dietary Supplement – The FD&C defines dietary supplement as a product that contains a “dietary ingredient” and is intended to supplement the diet. The “dietary ingredients” in these products may include 1) vitamins, 2) minerals, 3) herbs or other botanicals, 4) amino acids, 5) substances found in the diet (such as enzymes and edible organ tissues and glandulars, and 6) concentrates,metabolites, consitituents, extracts, or combinations of the substances identified in 1-5. Dietary supplements may be found in many forms such as tablets, capsules, softgels, gelcaps, liquids, or powders. Dietary supplements are regulated as foods, rather than drugs, for purposes of the FD&C Act and their labeling must identify them as a dietary supplement.
Dietary Supplement Claims - Under the FD&C, dietary supplements may bear a claim 1) regarding a benefit related to a classical nutrient deficiency, provided the claim discloses the prevalence of such disease in the United States, 2) describing the role of a nutrient or dietary ingredient intended to affect the structure of function in humans, 3) characterizing the documented mechanism by which a nutrient or dietary ingredient acts to maintain such structure or function, or 4) describing general well-being from consumption of a nutrient or dietary ingredient.
Clinical Investigation – IND regulations define clinical investigation as an “experiment in which a drug is administered or dispensed to, or used involving, one or more human subjects. For the purposes of the IND regulations, an experiment is any use of a drug [whether approved or unapproved] except for the use of a marketed drug in the course of medical practice.
Research Studies that Require an IND
- The research involves a drug as defined in section 201(g)(1) of the FD&C Act.
- The research is a clinical investigation as defined in the IND regulations.
- The clinical investigation is not otherwise exempt from the IND requirements.
Research Studies that are Exempt from an IND
FDA regulations describe two categories of clinical investigations that are exempt from the IND requirements in part 312, provided the criteria for exemption are met.
Research Involving Marketed Drug Products
A clinical investigation of a marketed drug is exempt from the IND requirements if all of the criteria for an exemption in § 312.2(b) are met:
- The drug product is lawfully marketed in the United States.
- The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication and there is no intent to use it to support any other significant change in the labeling of the drug.
- In the case of a prescription drug, the investigation is not intended to support a significant change in the advertising for the drug.
- The investigation does not involve a route of administration, dose, patient population, or other factor that significantly increases the risk (or decreases the acceptability of the risk) associated with the use of the drug product (21 CFR 312.2(b)(1)(iii)).
- The investigation is conducted in compliance with the requirements for review by an IRB (21 CFR part 56) and with the requirements for informed consent (21 CFR part 50).
- The investigation is conducted in compliance with the requirements of § 312.7 (i.e., the investigation is not intended to promote or commercialize the drug product).
Bioavailability or Bioequivalence Studies in Humans
FDA regulations describe criteria under which bioavailability or bioequivalence (BA/BE) studies using unapproved versions of approved drug products can be conducted without submission of an IND (21 CFR 320.31(b) and (d)). Although these regulations are intended to facilitate development of generic drugs, a planned BA/BE study need not be intended for that purpose to be exempt from the IND regulations. A BA/BE study in humans does not require an IND if all of the following conditions are met:
- The drug product does not contain a new chemical entity (21 CFR 314.108), is not radioactively labeled, and is not cytotoxic.
- The dose (single dose or total daily dose) does not exceed the dose specified in the labeling of the approved version of the drug product.
- The investigation is conducted in compliance with the requirements for review by an IRB (21 CFR part 56) and with the requirements for informed consent (21 CFR part 50).
- The sponsor meets the requirements for retention of test article samples (21 CFR 320.31(d)(1)) and safety reporting (21 CFR 320.31(d)(3)).
Inquiries Concerning the Application of the IND Requirements
If an investigator is uncertain about the applicability of an IND, we recommend that the investigator contact the appropriate review division (i.e., for the therapeutic area being studied) in the appropriate FDA center for a pre-IND submission consultation.
- How do I know if my product is regulated by the FDA simplified flow chart.
- Organizational charts listing the CDER review divisions and their telephone numbers are available athttps://www.fda.gov/media/77025/download.
- If the relevant review division is not known, we recommend you contact CDER’s Division of Drug Information (druginfo@fda.hhs.gov) or CBER’s Division of Manufacturer’s Assistance and Training (matt@cber.fda.gov), Office of Communication, Outreach and Development.
20.1 Use of Approved Drugs for Off-Label Indications
Investigational Purposes
If an investigator is using an approved drug in the context of a study protocol (i.e., to gather data for the purpose of changing the drug’s labeling) then the investigation would be subject to IRB review and approval, including informed consent requirements and may also be subject to regulation by the FDA.
Non-Investigational Purposes
If a physician prescribes a marketed drug for an indication not in the approved labeling, s/he has the responsibility to be well informed about the product and to base its use on a firm scientific rationale and on sound medical evidence, and to maintain records of the products and its effects. However, use of a product in this manner as part of the practice of medicine, where no data will be gathered for research purposes, does not require the approval of the IRB.
20.2 Expanded Access of Investigational Drugs (Compassionate Use)
Expanded access, sometimes referred to as “compassionate use," is the use outside of a clinical trial of an investigational medical product (i.e., one that has not been approved by FDA).
The FDA has an extensive website with information about how to access compassionate use drugs and devices. Wherever possible, use of an investigational medical product by a patient as part of a clinical trial is preferable because clinical trials can generate data that may lead to the approval of products and, consequently, to wider availability. However, when patient enrollment in a clinical trial is not possible (e.g., a patient is not eligible for any ongoing clinical trials, or there are no ongoing clinical trials), patients may be able to receive the product, when appropriate, through expanded access.
Requirements for All Expanded Access Uses
Under the Federal Food, Drug, and Cosmetic Act (FD&C Act), a patient may seek individual patient expanded access to investigational products for the diagnosis, monitoring, or treatment of a serious disease or condition if the following conditions are met.
- The patient and a licensed physician are both willing to participate.
- The patient's physician determines that there is no comparable or satisfactory therapy available to diagnose, monitor, or treat the patient’s disease or condition.
- That the probable risk to the person from the investigational product is not greater than the probable risk from the disease or condition.
- FDA determines that there is sufficient evidence of the safety and effectiveness of the investigational product to support its use in the particular circumstance;
- FDA determines that providing the investigational product will not interfere with the initiation, conduct, or completion of clinical investigations to support marketing approval;
- The sponsor (generally the company developing the investigational product for commercial use) or the clinical investigator (or the patient’s physician in the case of a single patient expanded access request) submits a clinical protocol (a document that describes the treatment plan for the patient) that is consistent with FDA’s statute and applicable regulations for INDs or investigational device exemption applications (IDEs), describing the use of the investigational product; and
- The patient is unable to obtain the investigational drug under another IND or to participate in a clinical trial.
Also under the FD&C Act statute, a sponsor or a physician may submit a protocol intended to provide widespread access to an investigational product for multiple patients. In this scenario, FDA will permit the investigational product to be made available under a treatment IND if certain criteria are met.
Ensuring patient safety is a priority - FDA must determine that the potential patient benefit justifies the potential risk of the expanded access use of the investigational drug, and that the potential risk is not unreasonable in the context of the disease or condition to be treated. Even with safeguards, there may be significant unknowns about safety and effectiveness.
Expanded Access Categories
21 CFR part 312 subpart I provides general requirements, describes criteria that must be met to authorize expanded access, lists requirements for expanded access submissions, and describes safeguards that will protect patients and preserve the ability to develop meaningful data about the use of the investigational product.
Under FDA’s current regulations for investigational drugs and biologics, there are three categories of expanded access:
Expanded Access for Individual Patients
- Individual Patient Expanded Access IND (Single Patient IND)
Access to an investigational drug (including a biologic) for use by a single patient submitted as a protocol under a new IND. The investigational product may or may not be under development. Unless FDA notifies the sponsor that treatment may begin earlier, there is a 30-day waiting period before treatment with the drug may begin.
- Individual Patient Expanded Access Protocol (also referred to as Single Patient Protocol)
Access to an investigational drug (including a biologic) for use by a single patient submitted as a new protocol to an existing IND by the sponsor of the existing IND. Typically, several patients may follow the same protocol. The investigational product may or may not be under development. There is no 30- day waiting period before treatment with the investigational product may begin, but the protocol must be received by FDA and have approval by an Institutional Review Board (IRB) before treatment may begin.
- Individual Patient Access in an Emergency (also known as emergency expanded access use and emergency access)
Please note that Emergency INDs and protocols are a subset of Individual Patient Access.
(1) Emergency IND: Individual Patient Access IND for Emergency use: Access to an investigational drug (including a biologic) for use by a single patient in an emergency situation (i.e., a situation that requires a patient to be treated before a written submission can be made) submitted as a protocol under a new IND. Treatment is initially requested and authorized by telephone or other rapid means of electronic communication, and may start immediately upon FDA authorization. The written submission (i.e., the individual patient expanded access IND) must be submitted within 15 business days of the telephone authorization.
(2) Emergency Protocol: Individual Patient Expanded Access Protocol for Emergency Use: Access to an investigational drug (including a biologic) for use by a single patient in an emergency situation (i.e., a situation that requires a patient to be treated before a written submission can be made) submitted as a new protocol to an existing IND by the sponsor of the existing IND. Treatment is initially requested and authorized by telephone or other rapid means of communication, and treatment may start immediately upon FDA authorization. The written submission (i.e., the individual patient expanded access protocol) must be submitted within 15 business days of the telephone authorization.
In an emergency situation where there is not sufficient time to secure IRB review prior to beginning treatment, the emergency use of the investigational drug must be reported to the IRB within 5 working days, as required under 21 CFR 56.104(c). See additional guidance on Emergency Use of an Investigational Drug or Biologic.
Expanded Access for Intermediate-Size Patient Populations
- Intermediate-size Patient Population Expanded Access IND
Access to an investigational drug (including a biologic) for use by more than one patient, but generally fewer patients than are treated under a typical treatment IND or protocol, submitted as a protocol under a new IND. The investigational product may or may not be under development for marketing. Unless FDA notifies the sponsor that treatment may begin earlier, there is a 30-day waiting period before treatment may begin.
- Intermediate-size Patient Population Expanded Access Protocol
Access to an investigational drug (including a biologic) for use by more than one patient, but generally fewer patients than are treated under a typical treatment IND or protocol, submitted as a protocol to an existing IND by the sponsor of the existing IND. The investigational product may or may not be under development for marketing. There is no 30-day waiting period before treatment with the investigational product may begin, but the protocol must be received by FDA and have IRB approval before treatment may begin.
An intermediate-size patient population protocol may also be requested to allow access to treatment with an approved drug (including a biologic) or a related product that is not available through marketing channels because of failure to meet the conditions of approval or a drug shortage, provided the drug and the patient meet the general criteria for expanded access as well as the criteria specific to use in an intermediate-size patient population.
Expanded Access for Widespread Use
- Treatment IND:
Access to an investigational drug (including a biologic) for treatment use by a large (widespread) population, submitted as a protocol under a new IND. The investigational product must be under active development for marketing. Unless FDA notifies the sponsor that treatment may begin earlier, there is a 30-day waiting period before treatment may begin.
- Treatment Protocol:
Access to an investigational drug (including a biologic) for treatment use by a large (widespread) population, submitted as a protocol to an existing IND by the sponsor of the existing IND. The investigational product must be under development for marketing. Unlike other access protocols submitted to existing INDs, there is a 30-day waiting period before treatment may begin, unless FDA notifies the sponsor that treatment may begin earlier.
Consider Investigational Product Availability and Costs
- The medical product company must agree to provide the investigational drug for expanded access use. FDA cannot require a company to provide an investigational drug for expanded access use to proceed.
- A company may decide to turn down a request if, for example it is not able or willing to provide access to an investigational drug outside of clinical trials intended to support marketing approval.
- In some cases, patients may have to pay for using the investigational drug and/or for medical care associated with the use of the investigational drug. In others, pharmaceutical companies may elect not to charge.
IRB Review
If a researcher wishes to treat a patient who is eligible to receive an expanded access investigational drug, the IRB must be notified immediately. The IRB will need to know under which category the use falls (individual, intermediate, or widespread as listed above), the timeline for treatment, and be provided with a protocol (which can be from the sponsor or investigator written), a consent (which can be developed from either the sponsor template or the IRB template), completion of the appropriate FDA IND submission forms and the IND application number if the PI has obtained it already.
To Allow for IRB Chairperson Concurrence (versus convened meeting) for an Individual Patient IND
A physician submitting an individual patient expanded access IND using Form FDA 3926, may select the appropriate box on that form to request authorization to obtain concurrence by the IRB chairperson or by a designated IRB member before the treatment use begins, in lieu of obtaining IRB review and approval at a convened IRB meeting at which a majority of the members are present. In order to utilize the expedited Chair review process, PI’s must complete Form FDA 3926 and check box 10.b. under 21 CFR 56.105.
20.3 Food and Food-Derived Products, Spices/Herbs, or Dietary Supplements
Drug or Disease Purpose vs. Structure/Function
FDA has further defined “disease claims” as including any statement that a product:
- Has an effect on the characteristic signs or symptoms of a specific disease or classic diseases;
- Belongs to a class of products that is intended to diagnose, mitigate, treat, cure, or prevent a disease or class of diseases;
- Augments a particular therapy or drug action that is intended to diagnose, mitigate, treat, cure, or prevent a disease or class of diseases;
- Otherwise suggests an effect on a disease or diseases.
Acceptable structure/function claims | Disease or Drug Claims |
Mild memory loss associated with aging | Alzheimer’s disease or senile dementias in the elderly |
Noncystic acne | Cystic acne |
To help manage mild mood changes, cramps, and edema associated with the menstrual cycle | Depression associated with the menstrual cycle |
Maintaining cholesterol levels in people with normal levels | Lowers cholesterol |
Inquiries Concerning the Application of the IND Requirements
Organizational charts listing the CDER review divisions and their telephone numbers are available at https://www.fda.gov/media/77025/download.
- If the relevant review division is not known, we recommend you contact CDER’s Division of Drug Information (druginfo@fda.hhs.gov) or CBER’s Division of Manufacturer’s Assistance and Training (matt@cber.fda.gov), Office of Communication, Outreach and Development.
20.4. Controlled Substances Used in Research
New 04/01/21
Controlled substances (CS) are often used in the animal research settings for anesthesia, analgesia, sedation, or euthanasia. They can also be used in human subject substance abuse protocols. Controlled substances are drugs which are regulated by the Drug Enforcement Administration (DEA) because of potential for abuse.
Each investigator or research location which uses CS must complete a registration with the DEA and obtain approval from the UVM Controlled Substances Committee (CSC) following the University Operating Policy – Controlled Substances in Research.
21. Investigational Devices
Revised 06/15/21
Investigational devices are medical devices which are the object of clinical research to determine their safety or effectiveness. Studies undertaken to develop safety and effectiveness data for medical devices involving human participants must be conducted according to the requirements of the Investigational Device Exemption (IDE) regulations (21 CFR 812).
IRB Review
When the device study does not have an Investigational Device Exemption, the IRB must determine the type of investigational device being used based upon risk. There are three types of device studies described under this regulation: significant risk (SR) device studies, non-significant (NSR) device studies, and exempt studies.
A significant risk (SR) device means an investigational device that:
- Is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a participant;
- Is purported or represented to be for use supporting or sustaining human life and presents a potential for serious risk to the health, safety, or welfare of a participant;
- Is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a participant; or
- Otherwise presents a potential for serious risk to the health, safety, or welfare of a participant.
A SR device must follow all the IDE Regulations at 21 CFR 812 and the study must be conducted under an Investigational Device Exemption (IDE) application approved by the FDA before the investigation begins.
FDA considers studies of all significant risk devices to present more than minimal risk; thus, IRB review at a convened meeting is required for all studies involving significant risk devices. To determine if a research device presents significant or non-significant risks, the IRB will consider the device’s total risks following the definitions found below. The device’s risk will not be compared with the risks of alternative devices or procedures. If the research device is used in conjunction with a procedure that involves risk, the IRB will consider the combined risks. The IRB will need to consider if a participant is to undergo any additional procedures as part of the investigational study, thus increasing risk. Once a determination on the degree of risk is reached, the IRB will consider whether the study meets the regulatory criteria for approval.
The UVM IRB must review at a convened meeting a description of the device, reports of prior investigations conducted with the device, the proposed investigational plan, and participant selection criteria. The sponsor should provide the IRB with a risk assessment and the rationale used in making its determination.
Examples of significant risk devices: catheters (other than urological), ventilators, CPR devices, TMJ prostheses, stents, lithotripters, sutures and absorbable bandages/materials, ECT devices, extended wear contact lenses, pacemakers, contraceptive devices, most laser systems, and most hemodialysis systems.
A non-significant risk (NSR) device is any device that does not meet the definition of a SR device. It is the sponsor’s responsibility to provide the IRB with the following:
- their determination that the device is NSR,
- the reasons why it has come to this conclusion,
- information needed to allow the IRB to evaluate the risk of using the device in the proposed study,
- a description of the device,
- the protocol and any other information that the IRB requires
Studies involving a NSR need only to follow the abbreviated device regulations at 21 CFR 812.2(b).
At the time of its review the IRB must review the investigator/sponsor's NSR determination and either agree or disagree. To determine if a research device presents significant or non-significant risks; the IRB will consider the device’s total risks following the definitions found below. The device’s risk will not be compared with the risks of alternative devices or procedures. If the research device is used in conjunction with a procedure that involves risk, the IRB will consider the combined risks. Once a determination on the degree of risk is reached, the IRB will consider whether the study meets the regulatory criteria for approval.
Some studies involving non-significant risk devices may be categorized as not more than minimal risk studies and thus may be reviewed through the expedited review procedure established by the IRB.
NSR device studies do not have to have an IDE application approved by FDA.
The UVM IRB’s NSR determination is important because the UVM IRB serves as the FDA’s surrogate for review, approval, and continuing review of the NSR device. An NSR device study may start at the institution as soon as the IRB reviews and approves the study and without prior approval by FDA.
Examples of non-significant risk devices: most daily wear contact lenses, lens solutions, heel cups, antibacterial surgical garments, incontinence devices, Digital Mammography, and ultrasonic tooth cleaners.
An exempt investigational device study means, that with the exception of 21 CFR 812.119, the rest of the IDE regulations do not apply 21 CFR 812.2 (c)(link is external). These devices also do not require the FDA, the sponsor, the investigator, or the IRB to make an SR or NSR determination.
Exempted investigations include investigations involving one of the following:
- A device, other than a transitional device, in commercial distribution immediately before May 28, 1976, when used or investigated in accordance with the indications in labeling in effect at that time.
- A device, other than a transitional device, introduced into commercial distribution on or after May 28, 1976, that FDA has determined to be substantially equivalent to a device in commercial distribution immediately before May 28, 1976, and that is used or investigated in accordance with the indications in the labeling FDA reviewed under subpart E of part 807 in determining substantial equivalence.
- A diagnostic device, if the sponsor complies with applicable requirements in 809.10(c) and if the testing: (Comment: this is the most frequent type of exempted investigation submitted to the IRB.)
- Is noninvasive,
- Does not require an invasive sampling procedure that presents significant risk,
- Does not by design or intention introduce energy into a participant, and
- Is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic product or procedure.
- A device undergoing consumer preference testing, testing of a modification, or testing of a combination of devices if the device(s) are legally marketed device(s) (that is, the devices have an approved PMA, cleared Premarket Notification 510(k), or are exempt from 510(k)) AND if the testing is not for the purpose of determining safety or effectiveness and does not put participants at risk.
- A device intended solely for veterinary use.
- A device shipped solely for research on or with laboratory animals and labeled in accordance with 812.5(c).
- A custom device as defined in 812.3(b), unless the device is being used to determine safety or effectiveness for commercial distribution.
The IRB does not need to decide whether the study poses a significant risk or nonsignificant risk. However, the IRB must still review the study in accordance with the IRB regulations before the investigation may begin.
Some studies involving exempt investigations may be categorized as not more than minimal risk studies and thus may be reviewed through the expedited review procedure established by the IRB.
21.1. Expanded Access of Investigational Devices
When a patient has a serious or life-threatening condition that is not addressed by current approved treatments, options may exist to use an investigational medical device (i.e., one that has not been approved or cleared by FDA) to treat the patient.
Normally, such investigational devices with significant risks may only be used on human subjects through an FDA-approved clinical trial for which an investigational device exemption (IDE) allows the investigational device to be used in a clinical study. However, there are circumstances under which a health care provider may use an investigational device outside of a clinical study to save the life of a patient or to help a patient suffering from a serious disease or condition for which no other alternative therapy exists.
The use of an investigational device outside of a clinical trial for treatment of a patient is called “expanded access.” If enrollment in an existing clinical trial protocol is not possible (e.g., a patient is not eligible for any ongoing clinical trials, or there are no ongoing clinical trials to address the patient’s condition), patients/physicians have the potential to receive expanded access to investigational devices under one of three alternative mechanisms: emergency use, expanded access use, and treatment use. The FDA has an extensive website with information on process and how to apply.
Emergency Use
Emergency use is the use of an investigational device in an emergency situation. It is intended to provide patients and physicians with access to devices intended to treat life-threatening or serious diseases or conditions when there is no available alternative and no time to obtain FDA approval. Criteria for emergency use are:
- The patient has a life-threatening or serious disease or condition that needs immediate treatment;
- No generally acceptable alternative treatment for the conditions exists; and
- Because of the immediate need to use the device, there is no time to use existing procedures to obtain FDA approval for the use.
See Emergency Use of an Investigational Drug or Biologic or Investigation Device section for information regarding process for informing the IRB.
Expanded Access Use
FDA recognizes that there are circumstances in which an investigational device is the only option available for a patient faced with a serious or life-threatening disease or condition.
The use provision provides a path to accessing investigational devices that have not received FDA approval or clearance for patients for whom the treating physician believes the device may provide a benefit in treating and/or diagnosing their disease or condition. use can be for devices that are being studied in a clinical trial under an IDE for patients who do not meet the requirements for inclusion in the clinical investigation but for whom the treating physician believes the device may provide a benefit in treating and/or diagnosing their disease or condition. It can also be used for devices that are not being studied in a clinical investigation (i.e., an IDE for the device does not exist). This provision is typically approved for individual patients but may be approved to treat a small group. Criteria for compassionate use are:
- The patient has a life-threatening or serious disease or condition; and
No generally acceptable alternative treatment for the condition exists.
Prior full committee IRB review and approval is required.
Treatment Use
An approved IDE specifies the maximum number of clinical sites and the maximum number of human subjects that may be enrolled in the study. During the course of the clinical trial, if the data suggest that the device is effective, then the trial may be expanded to include additional patients with life-threatening or serious diseases. This is called treatment use. Criteria for treatment use are:
- The device is intended to treat or diagnose a serious or immediately life-threatening disease or condition;
- There is no comparable or satisfactory alternative device available to treat or diagnose the disease or condition in the intended patient population;
- The device is under investigation in a controlled clinical trial for the same use under an approved IDE, or all clinical trials have been completed; and
- The sponsor of the controlled clinical trial is pursuing marketing approval/clearance of the investigational device with due diligence.
Prior full committee IRB review and approval is required.
22. Humanitarian Use Device (HUD) Designation and Humanitarian Device Exemptions (HDE)
Definitions 21 CFR 812.3
Humanitarian Use Device (HUD) – a medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in not more than 8,000 individuals in the United States per year.
Humanitarian Device Exemption (HDE) – is a marketing application for a HUD which is exempt from the effectiveness requirements and is subject to certain profit and use conditions.
An HDE application is not required to contain the results of scientifically valid clinical investigations demonstrating that the device is effective for its intended purpose. The application, however, must contain sufficient information for FDA to determine that the device does not pose an unreasonable or significant risk of illness or injury, and that the probable benefit to health outweighs the risk of injury or illness from its use, taking into account the probable risks and benefits of currently available devices or alternative forms of treatment. Additionally, the applicant must demonstrate that no comparable devices are available to treat or diagnose the disease or condition, and that they could not otherwise bring the device to market.
An approved HDE authorizes marketing of the HUD. However, a HUD may only be used in a facility after an IRB has approved their use in that facility.
IRB Requirements
HUDs are typically used for clinical purposes not research purposes. However, the regulations require prior IRB review and approval.
Per 21 CFR 814.124, “a HUD may be administered only if such use has been approved by the IRB located at the facility.” “If, however, a physician in an emergency situation determines that approval from an IRB cannot be obtained in time to prevent serious harm or death to a patient, a HUD may be administered without prior approval by the IRB located at the facility.” In such an emergency situation, the physician shall, within 5 days after the use of the device, provide written notification to the chairman of the IRB of such use. Such written notification shall include the identification of the patient involved, the date on which the device was used, and the reason for the use. In these situations, clinicians must follow the procedures for Emergency Use of Investigational Drug or Device, Section 23.
Initial proposed use of a HUD will be reviewed by a convened meeting of the IRB in accordance with 21 CFR 56.111.
FDA allows for continuing approval of the HUD to be conducted by expedited review as it is a legally marketed device and no safety and effectiveness information is being collected systematically as would be typical for a clinical trial.
Consent
The IRB does not require development of a written informed consent for review and approval for these clinical situations. Clinicians should provide patients with information about the device and document patient’s consent for device placement in the medical chart similar to other clinical procedures.
If the HUD is being studied in a clinical trial setting, prior written research consent is required following standard procedures for human subjects research.
Clinician Responsibilities
- Clinician is responsible to obtain IRB approval prior to use.
- Ensures that the HDE exists for the use of the HUD and that the proposed use meets the HDE requirements.
- Submits basic written plan for use of the HUD, HUD manufacturer’s product labeling, clinical brochure and/or other pertinent manufacturer information materials, the FDA HDE approval letter
- Submit documentation to the IRB for continuing review.
- Current FDA-approved HUD manufacturer’s product labeling
- Submit adverse events following the IRB criteria. This is in addition to the FDA and/or manufacturer reporting requirements.
- Submit information to the FDA and the IRB whenever a HUD may have caused or contributed to a death or serious injury.
IRB Responsibilities
- The IRB will convene a full meeting to review use of the HUD. The IRB will
- ensure that the proposed use is within the FDA-approved indication and the use of the device does not exceed the scope of the FDA’s approval and
- verify that the device does not pose an unreasonable risk of illness or injury to the recipient, and that the probable benefit outweighs the risks from use of the device.
The IRB does not have to review and approve each individual use of the HUD. The IRB may approve the use of the device in general, for groups of patients clinically appropriate for the device’s intended use.
- Provide continuing review by expedited review at least annually
- Review adverse events and unanticipated problems to subjects or others related to the use of the device.
23. Emergency Use of an Investigational Drug or Biologic or Investigation Device
Revised: 2/22/22
Emergency use is defined as the use of a test article (investigational drug or biological product or investigational device) on a human subject in a life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval." 21 CFR 56.102(d)
Generally, IRB approval is required prior to conducting human subject research. However, an exception to this is the one-time use of an investigational drug or device (test article) for a single participant in a life-threatening (emergency use) situation.
An emergency use of a test article is exempt from prior IRB review and approval, provided that the emergency use of a test article is reported to the IRB within 5 working days of the date of the emergency use.
Any subsequent use of the test article at UVM is subject to IRB review and approval. Only one emergency use of the test article is permitted and any subsequent use needs to be done under an IRB approved protocol. However, the FDA acknowledges that it would be inappropriate to deny emergency treatment to a second individual if the only obstacle is that the IRB has not had sufficient time to convene a meeting to review the issue. (FDA Information Sheet, 2003 Update)
Contact RPO for an Emergency Use of an Investigational Drug, Biologic, or Device Certification of Compliance Form. The IRB Chair or designated IRB member will review the submission.
Criteria for Emergency Use
All the following must be satisfied
- Existence of a life-threatening/severely debilitating condition where no standard acceptable treatment is available
- No current IRB approved protocol covering the situation and no time to obtain prior FDA and IRB approval
- Availability of an investigational agent or device which in the opinion of the physician might be beneficial, and
- Availability of an investigational agent or device from a sponsor or elsewhere.
- The Emergency Use of a Test Article is not a systematic investigation designed to develop or contribute to generalizable knowledge.
If the proposed use meets this definition, the investigator must contact the IRB office to request concurrence with administration of the test article.
During Normal Business Hours
Contact the RPO Office at 656-5040
Outside Normal Business Hours Use Provider Access System to Contact One of the Chairs
Click here to find the current IRB Chairs
Consent Requirement
The process of informed consent must meet FDA requirements [21 CFR 50.25]. The investigator is required to obtain legally effective informed consent of the subject or the subject’s legally authorized representative, using an appropriate consent document. Whenever possible, a copy of the consent form to be used is requested in advance (generally a standard form exists and the IRB does not require that it be put into the usual institutional format for an emergency use). The IRB has a consent template located on its forms page for use.
Exception from Informed Consent Requirement
Informed consent of the subject or the subject’s legally authorized representative is required, unless both the investigator and a physician (not otherwise participating in the investigation) certify in writing that:
- the patient is confronted with a life-threatening situation;
- informed consent cannot be obtained from the patient (because patient cannot communicate or is incompetent to give consent);
- consent cannot be obtained from the legally authorized representative (unavailable or unknown); and
- no alternative approved treatment/therapy is available that provides an equal or greater likelihood of saving the patient’s life. (21 CFR 50.23(2)).
Emergency Use with Drugs and Biologics
The investigator must submit the following materials to the IRB within five (5) working days, following the use of the test article:
- information about the patient
- indication of the life-threatening or severely debilitating nature of the situation
- explanation as to why this drug or treatment was necessary
- and, if the emergency use occurred without obtaining prior informed consent, Section D on this form must also be completed: Independent Physician Certification - Emergency Use of a Test Article Without Informed Consent
- Written permission from the manufacturer for the use of the test article under their IND. Generally the investigator will contact the manufacturer and determine if the drug or biologic can be made available for the emergency use under the company’s IND. If the company only allows cross-referencing to their IND, declines permission or cannot be reached, the investigator should contact the FDA for authorization of the shipment of the drug in advance of the IND submission. In such a case the FDA may authorize shipment of the test article in advance of the IND submission. The IRB will request that the investigator contact the FDA to obtain an Emergency Use IND.
- A copy of the signed Consent Form.
Emergency Use with Devices
The investigator must submit the following materials to the IRB within five (5) working days following the procedure:
- information about the patient
- indication of the life-threatening or severely debilitating nature of the situation
- explanation as to why this device was necessary
- and, if the emergency use occurred without obtaining prior informed consent, Section D on this form must also be completed: Independent Physician Certification - Emergency Use of a Test Article Without Informed Consent
- Written permission from the manufacturer for the use of the test article under their IDE. Generally the investigator will contact the manufacturer and determine if the device can be made available for the emergency use under the company’s IDE. The IRB will request that the investigator contact the FDA to obtain an IDE. If the company only allows cross-referencing to their IDE, declines permission or cannot be reached or an IDE does not exist, the FDA expects the investigator to:
- Determine whether the criteria for emergency use have been met;
- Assess the potential for benefits from the unapproved device and to have substantial reason to believe that benefits exist;
- Assure that the decision of the investigator that an emergency exists is not based solely on the expectation that IDE approval procedures may require more time than is available.
- Obtain an independent assessment by an uninvolved physician.
In addition, if the device is used and there is no IDE:
- The use must be reported to the FDA within 5 working days (to CDRH). This report should contain a summary of the conditions constituting the emergency, patient outcome information, and the patient protection measures that were followed.
- Copy of signed Consent Form.
FDA Emergency use requests
- For investigational biological products regulated by CBER, call 301-827-1800.
- For all other investigational drugs, call 301-796-3400.
- After working hours, call FDA's Office of Emergency Operations at 1-866-300-4374 or 301-796-8240.
- CDER website - including Emergency Use
24. Subjects Vulnerable to Coercion or Undue Influence
Revised: 2/10/2023
45 CFR 46.111(b)
Vulnerability to coercion or undue influence references limitations to a person’s ability to provide informed consent to participate in research. These limitations could be due to a person’s current circumstances (in the case of prisoners or the educationally or economically disadvantaged), or due to a temporary or permanent lack of capacity (in the case of the children and individuals with impaired decision-making capacity).
The Common Rule changed the categories of subjects that are classified as vulnerable to coercion or undue influence. In addition to replacing the “mentally disabled” with the more accurate and sensitive “individuals with impaired decision-making capacity,” the “handicapped” and “pregnant women” have been removed from all lists of vulnerable categories of subjects.
When making an assessment, the IRB will take into account the purposes of the research and the setting in which the research will be conducted. The IRB should be particularly cognizant of the special problems of research that involves the category of subjects who are vulnerable to coercion or undue influence, such as
- Children
- Prisoners
- Individuals with impaired decision-making capacity
- Economically or educationally disadvantaged individuals
Additional populations not specifically discussed within the regulations but for which additional unspecified safeguards could be required by the IRB, are listed below.
1. Students/employees;
2. Terminally ill patients;
3. Subjects with drug and/or alcohol addictions;
4. Subjects with other disabilities; or
5. Non-English speaking subjects.
The IRB reviews research involving vulnerable populations according to applicable requirements and guidelines and makes determinations using the IRB reviewer checklist. If the research includes a vulnerable population that does not have additional protections specified in the federal regulations, the board will evaluate the research proposal to ensure that precautions are taken to protect the participants.
Depending on the research, exclusion of any of the above populations might be construed as unfair and attempts should be made to include these populations, with appropriate protections, if they are applicable to the research question. If the protocol is records or specimen collection only and the vulnerable population cannot be identified or there is no risk to the vulnerable subject and they should not be listed as targeted subjects on your protocol.
24.1 Pregnant Women, Fetuses, Neonates of Uncertain Viability and Non-Viable Neonates 45 CFR 46 Subpart B
Revised 08/25/22
In reviewing Research that involves Pregnant Women, human Fetuses or Neonates, the UVM IRB shall ensure the research complies with the applicable requirements of 45 CFR Part 46, Subpart B.
Note: Subpart A, under the Revised Common Rule, removed Pregnant Women from the list of vulnerable populations. Subpart B was unchanged, so the same protections stand.
§46.202 Definitions
(a) Dead fetus - a fetus that exhibits neither heartbeat, spontaneous respiratory activity, spontaneous movement of voluntary muscles, nor pulsation of the umbilical cord.
(b) Delivery - a complete separation of the fetus from the woman by expulsion or extraction or any other means.
(c) Fetus - the product of conception from implantation until delivery.
(d) Neonate - a newborn.
(e) Nonviable neonate - a neonate after delivery that, although living, is not viable.
(f) Pregnancy - encompasses the period of time from implantation until delivery. A woman shall be assumed to be pregnant if she exhibits any of the pertinent presumptive signs of pregnancy, such as missed menses, until the results of a pregnancy test are negative or until delivery.
(g) Secretary - the Secretary of Health and Human Services and any other officer or employee of the Department of Health and Human Services to whom authority has been delegated.
(h) Viable, as it pertains to the neonate - means being able, after delivery, to survive (given the benefit of available medical therapy) to the point of independently maintaining heartbeat and respiration. The Secretary may from time to time, taking into account medical advances, publish in the FEDERAL REGISTER guidelines to assist in determining whether a neonate is viable for purposes of this subpart. If a neonate is viable then it may be included in research only to the extent permitted and in accordance with the requirements of subparts A and D of this part.
§46.204 Research involving pregnant women or fetuses
Research studies involving pregnant women or fetuses can be approved by the IRB if the following requirements of the federal regulations are satisfied 46.204:
The criteria for review will depend on the risk level (as determined by the IRB) and the applicability of federal regulations, as follows:
Non-Federally Regulated Minimal Risk Research (Expedited Review) -
The research must meet the following criteria, and no other determinations are necessary in terms of Pregnant Women and Fetuses:
- The research is NOT conducted, funded, or otherwise subject to regulation by DHHS, Environmental Protection Agency (EPA), or Veterans Administration (VA).
- The research involves no more than Minimal Risk to pregnant women and fetuses.
- Minimal Risk - the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life of the general population or during the performance of routine physical or psychological examinations or tests
- The research is not funded by Department of Defense or does not involve interventions/invasive procedures to the woman or fetus and does not involve fetuses or neonates as subjects.
More than Minimal Risk Studies (Full IRB Review), and Federally Regulated Research -
The research must meet ALL the following additional protection requirements:
(a) Where scientifically appropriate, preclinical studies, including studies on pregnant animals, and clinical studies, including studies on nonpregnant women, have been conducted and provide data for assessing potential risks to pregnant women and fetuses;
(b) The risk to the fetus is caused solely by interventions or procedures that hold out the prospect of direct benefit for the woman or the fetus; or, if there is no such prospect of benefit, the risk to the fetus is not greater than minimal and the purpose of the research is the development of important biomedical knowledge which cannot be obtained by any other means;
(c) Any risk is the least possible for achieving the objectives of the research;
(d) If the research holds out the prospect of direct benefit to the pregnant woman, the prospect of a direct benefit both to the pregnant woman and the fetus, or no prospect of benefit for the woman nor the fetus when risk to the fetus is not greater than minimal and the purpose of the research is the development of important biomedical knowledge that cannot be obtained by any other means, her consent is obtained in accord with the informed consent provisions of subpart A of this part;
(e) If the research holds out the prospect of direct benefit solely to the fetus then the consent of the pregnant woman and the father is obtained in accord with the informed consent provisions of subpart A of this part, except that the father's consent need not be obtained if he is unable to consent because of unavailability, incompetence, or temporary incapacity or the pregnancy resulted from rape or incest.
(f) Each individual providing consent under paragraph (d) or (e) of this section is fully informed regarding the reasonably foreseeable impact of the research on the fetus or neonate;
(g) For children as defined in §46.402(a) who are pregnant, assent and permission are obtained in accord with the provisions of subpart D of this part;
(h) No inducements, monetary or otherwise, will be offered to terminate a pregnancy;
(i) Individuals engaged in the research will have no part in any decisions as to the timing, method, or procedures used to terminate a pregnancy; and
(j) Individuals engaged in the research will have no part in determining the viability of a neonate.
§46.205 Research involving neonates
Neonates of uncertain viability and nonviable neonates may be involved in research if all of the following conditions are met:
- Where scientifically appropriate, preclinical, and clinical studies have been conducted and provide data for assessing potential risks to neonates.
- Each individual providing consent is fully informed regarding the reasonably foreseeable impact of the research on the neonate.
- Individuals engaged in the research will have no part in determining the viability of a neonate.
- The requirements of paragraph (b) or (c) of this section have been met as applicable.
(b) Neonates of uncertain viability. Until it has been ascertained whether or not a neonate is viable, a neonate may not be involved in research covered by this subpart unless the following additional conditions have been met:
(1) The IRB determines that:
(i) The research holds out the prospect of enhancing the probability of survival of the neonate to the point of viability, and any risk is the least possible for achieving that objective, or
(ii) The purpose of the research is the development of important biomedical knowledge which cannot be obtained by other means and there will be no added risk to the neonate resulting from the research; and
- (2) The legally effective informed consent of either parent of the neonate or, if neither parent is able to consent because of unavailability, incompetence, or temporary incapacity, the legally effective informed consent of either parent's legally authorized representative is obtained in accord with subpart A of this part, except that the consent of the father or his legally authorized representative need not be obtained if the pregnancy resulted from rape or incest.
(c) After delivery nonviable neonates may not be involved in research covered by this subpart unless all of the following additional conditions are met:
- Vital functions of the neonate will not be artificially maintained;
- The research will not terminate the heartbeat or respiration of the neonate;
- There will be no added risk to the neonate resulting from the research; and
- The purpose of the research is the development of important biomedical knowledge that cannot be obtained by other means; and
- The legally effective informed consent of both parents of the neonate is obtained in accord with subpart A of this part, except that the waiver and alteration provisions of §46.116(c) and (d) do not apply. However, if either parent is unable to consent because of unavailability, incompetence, or temporary incapacity, the informed consent of one parent of a nonviable neonate will suffice to meet the requirements of this paragraph (c)(5), except that the consent of the father need not be obtained if the pregnancy resulted from rape or incest. The consent of a legally authorized representative of either or both of the parents of a nonviable neonate will not suffice to meet the requirements of this paragraph (c)(5).
(d) Viable neonates
Newborns, after delivery, that are determined to be viable may be included in research only in accordance with the policies for including children in research under subpart A and D.
§46.206 Research involving, after delivery, the placenta, the dead fetus or fetal material.
Research involving, after delivery, the placenta; the dead fetus; macerated fetal material; or cells, tissue, or organs excised from a dead fetus, shall be conducted only in accord with any applicable Federal, State, or local laws and regulations regarding such activities.
If information associated with material described in this section is recorded for research purposes in a manner that individuals can be identified, directly or through identifiers linked to those individuals, those individuals are research subjects, and all pertinent regulations are applicable.
§46.207 Research not otherwise approvable which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of pregnant women, fetuses, or neonates.
The Secretary will conduct or fund research that the IRB does not believe meets the requirements of §46.204 or §46.205 only if:
(a) The IRB finds that the research presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of pregnant women, fetuses or neonates; and
(b) The Secretary, after consultation with a panel of experts in pertinent disciplines (for example: science, medicine, ethics, law) and following opportunity for public review and comment, including a public meeting announced in the FEDERAL REGISTER, has determined either:
(1) That the research in fact satisfies the conditions of §46.204, as applicable; or
(2) The following:
(i) The research presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of pregnant women, fetuses or neonates;
(ii) The research will be conducted in accord with sound ethical principles; and
(iii) Informed consent will be obtained in accord with the informed consent provisions of subpart A and other applicable subparts of this part.
24.2 Research Involving Prisoners - Subpart C
Revised 1.10.2023
Consult 45 CFR 46 Subpart C (Sections 301, 302, 303, 304, 305 and 306)
Prisoners, because of their incarceration, may be under constraints that could affect their ability to make a truly voluntary and un-coerced decision whether to participate as subjects in research. The purpose of this subpart is to provide additional safeguards for the protection of prisoners involved in research activities to which this subpart is applicable.
Scope
Prisoners as defined by HHS regulations under 46.303 are “any individual involuntarily confined or detained in a penal institution” and encompasses individuals sentenced to such an institution under criminal or civil statute, individuals detained in other facilities by virtue of statutes or commitment procedures which provide alternatives to criminal prosecution or incarceration in a penal institution, and individuals detained pending arraignment, trial, or sentencing.
Regulations do not automatically consider a person under a court order to be a “prisoner” under Subpart C. Study participants on parole or probation are NOT considered to be prisoners under Subpart C. Persons in post-release criminal justice halfway houses are presumptively NOT considered by OHRP to fit the Subpart C definition of prisoners. This is dependent upon whether detainment or confinement is voluntary.
IRB Review Requirements
Research involving prisoners will be reviewed by the convened UVM IRB committee which includes at least one member who is a (45 CFR 46.304) prisoner or a prisoner representative with appropriate background and experience to serve in that capacity. This includes initial review, continuing review, full-board modifications, and reportable unexpected or unanticipated problems.
- The VT Agency of Human Services and the University of Vermont (UVM) IRB have a reliance agreement in place that allows the AHS IRB to rely on the UVM IRB for collaborative projects led by a researcher at UVM involving prisoners. Researchers should contact the AHS IRB early in the review process to determine if they wish to review the protocol or rely on the UVM IRB review.
- New protocols should have a letter of support from the prison or correctional facility supporting the research protocol at their institution.
- Modifications that would otherwise be approvable by expedited review can be expedited (except for DoD funded studies) as long as the prisoner representative receives a copy of the modification and concurs that it does not adversely affect the prisoners.
Training Specific to Prisoner Populations
Additional training requirements through the CITI module are required for all key personnel working on research protocols involving more than minimal risk protocols that include a prisoner population. Please reference the CITI Program Training page on our website for additional information about adding this course to your profile.
Categories of Permitted Research with Prisoners (45 CFR 46.306)
The IRB may approve studies involving prisoners only if the research falls into one of the following four categories under Subpart C or meets a waiver of certain provisions for epidemiologic studies as described further below.
1) The research proposes to study the possible causes, effects, and processes of incarceration, and of criminal behavior. The study must be no more than minimal risk and no more than inconvenience to the participants. 46.306(a)(2)(i).
2) The research proposes to study prisons as institutional structures or to study prisoners as incarcerated persons. The study must be no more than minimal risk and no more than inconvenience to the participants. 46.306(a)(2)(ii)
Minimal risk is the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical, dental, or psychological examination of healthy persons.
3) The research proposes to study the conditions particularly affecting prisoners as a class. (For example: A vaccine trial and other research on hepatitis, which is much more prevalent in prisons than elsewhere or research on social and psychological problems such as alcoholism, drug addiction and sexual assaults.) Studies that fit into this category may only proceed after the Secretary of the Department of Health and Human Services has consulted with appropriate experts in penology medicine and ethics. The Secretary must also publish notice in the Federal Register of his/her intent to approve the research. 46.306(a)(2)(iii)
4) Research on practices, both innovative and accepted, that have the intent and reasonable probability of improving the health or well-being of the subject. In cases in which those studies require the assignment of prisoners in a manner consistent with protocols approved by the IRB to control groups that may not benefit from the research, the study may proceed only after the Secretary has consulted with appropriate experts, including experts in penology, medicine and ethics, and published notice in the Federal Register of his intent to approve such research. 46.306(a)(2)(iv)
There is a waiver of applicability of certain provisions [Federal Register, Vol. 68, No. 119, pp. 36929‐36931, Friday, June 20, 2003] of Subpart C when HHS is conducting or supporting epidemiologic research involving prisoners as subjects.
DHHS conducts or supports certain epidemiologic studies in which the purposes are as follows: (1) To describe the prevalence or incidence of a disease by identifying all cases, and (2) To study potential risk factor associations for a disease. For most such studies, the institutional review board (IRB) reviewing the study determines that the research at issue involves no more than minimal risk and no more than inconvenience to the subjects. The human participants in this type of public health research may include prisoners in the study population. State health agencies are most commonly the conduits for this type of research. Because certain epidemiologic studies conducted or supported by DHHS focus on a particular condition or disease that might affect prisoners as it would any other members of the general population, such studies do not meet any of the four categories of permissible research under subpart C, 45 CFR part 46.
Because these studies do not meet any of the four categories, the Secretary of DHHS may allow a waiver of the applicability of 46.305(a)(1) and 46.306(a)(2) in the conduct of certain important and necessary epidemiologic research on prisoners.
These studies (1) Sole purpose are (i) To describe the prevalence or incidence of a disease by identifying all cases, or (ii) To study potential risk factor associations for a disease, and (2) Where the institution is responsible for the conduct of the research certifies to OHRP, that the IRB approved the research and fulfilled its duties under 46.305(a)(2-7)(below) and determined and documented that (i) The research presents no more than minimal risk and no more than inconvenience to the prisoner-subject, and (ii) Prisoners are not a particular focus of the research.
Additional findings that the UVM IRB must make at time of review
Once the IRB has determined that the study is permissible with prisoners, the IRB must make the following additional findings under 45 CFR 46.305(a). These findings must be discussed and documented as part of the convened meeting minutes.
(A)(1) the research under review represents one of the categories of research permissible under Section 46.306(a)(2) above.
(2) any possible advantages accruing to the prisoner through his or her participation in the research, when compared to the general living conditions, medical care, quality of food, amenities and opportunity for earnings in the prison, are not of such a magnitude that his or her ability to weigh the risks of the research against the value of such advantages in the limited choice environment of the prison is impaired;
(3) the risks involved in the research are commensurate with risks that would be accepted by nonprisoner volunteers.
(4) procedures for the selection of subjects within the prison are fair to all prisoners and immune from arbitrary intervention by prison authorities or prisoners. Unless the principal investigator provides to the Board justification in writing for following some other procedures, control subjects must be selected randomly from the group of available prisoners who meet the characteristics needed for that particular research project;
(5) the information is presented in language which is understandable to the subject population.
(6) adequate assurance exists that parole boards will not take into account a prisoners participation in the research in making decisions regarding parole, and each prisoner is clearly informed in advance that participation in the research will have no effect on his or her parole; and
(7) where the Board finds there may be a need for follow-up examination or care of participants after the end of their participation, adequate provision has been made for such examination or care, taking into account the varying lengths of individual prisoners’ sentences, and for informing participants of this fact.
(B) the Board shall carry out such other duties as may be assigned by the Secretary.
(C) The institution shall certify to the Secretary, in such form and manner as the Secretary may require, that the duties of the Board under this section have been fulfilled.
What happens if a human subject becomes a prisoner while enrolled in a research study?
If a human subject involved in ongoing research becomes a prisoner during the course of the study, and the relevant research proposal was not reviewed and approved by the IRB in accordance with the requirements for research involving prisoners under subpart C of 45 CFR part 46, the investigator must promptly notify the IRB. All research interactions and interventions with, and obtaining identifiable private information about, the now-incarcerated prisoner-subject must be suspended immediately, except as noted below. Upon receipt of the investigator's report that a previously enrolled research subject has become a prisoner, if the investigator wishes to have the prisoner subject continue to participate in the research, the IRB must promptly re-review the proposal in accordance with the requirements of subpart C. If the proposal is federally supported, the institution(s) engaged in the research involving the prisoner subject must send a certification to OHRP and wait for a letter of authorization. Otherwise, the prisoner subject must stop participating in the research, except as noted below.
OHRP allows one important exception to the requirement that all research interactions or interventions with, and obtaining identifiable private information about, the now-incarcerated prisoner-subject must cease until the regulatory requirements for research involving prisoners are met. In special circumstances in which the investigator asserts that it is in the best interests of the subject to remain in the research study while incarcerated, the subject may continue to participate in the research until the requirements of subpart C are satisfied. The investigator must promptly notify the IRB of this occurrence, so that the IRB can re-review the study. Note that in these circumstances, some of the findings required by 45 CFR 46.305(a) may not be applicable; for example, the finding required under 45 CFR 46.305(a)(4) regarding the selection of subjects within the prison may not be applicable, if the subject was recruited outside of an incarcerated context. The IRB will need to document findings of non-applicability accordingly.
When preparing for a modification to your protocol to request the continuation of a research subject who has become a prisoner, it is unlikely that previous review of the research and the consent document contemplated the constraints imposed by incarceration. In this case, a modification that includes appropriate justification (benefits to the individual outweigh the additional risks due to incarceration) should be written including specific examples of benefits to the individual prisoner-subject. As part of the submission a consent addendum that the now prisoner-subject will need to sign in addition to the original consent must also be submitted for review and approval.
Note that prisoners cannot be involved in emergency research where the requirement for informed consent has been waived by the Secretary under the authority of 45 CFR 46.101(i).
Additional Federal Approval Requirements for Research Protocols Funded by HHS
If the research that involves prisoners is being funded by HHS, then in addition to the requirements specified above, the UVM IRB must prepare a prisoner certification letter to OHRP. The UVM IRB will certify it has made the seven findings required under 45 CFR 46.305(a), including the finding that the proposed research represents one of the permissible categories of research under 45 CFR 46.306(a)(2). The institution will send OHRP a completed Subpart C Certification Form, which includes the name and address of the institution and the relevant grant number to subpartc@hhs.gov.
Additionally, UVM will submit to OHRP a copy of the research proposal so OHRP can determine whether the proposed research involves one of the categories of research permissible under 45 CFR 46.306(a)(2), and if so, which one. The term "research proposal" includes:
- the IRB-approved protocol;
- any relevant HHS grant application or proposal;
- any IRB application forms required by the IRB; and
- any other information requested or required by the IRB to be considered during initial IRB review
24.3 Children
Research that is allowable with children is determined by the degree of risk involved. These categories of allowable research are:
1) No greater than minimal risk where adequate provisions are made for soliciting the assent of the children and the permission of their parents or guardians.
2) Research involving greater than minimal risk but presenting the prospect of direct benefit to the individual subjects, if the IRB finds that:
a. The risk is justified by the anticipated benefits to the subjects;
b. The relation of the anticipated benefit to the risk is at least as favorable to the subjects as that presented by available alternative approaches; AND,
c. Adequate provisions are made for soliciting the assent of the children and permission of their parents or guardians.
3) Research involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generalizable knowledge about the subject's disorder or condition, if the IRB finds that:
a. The risk represents a minor increase over minimal risk;
b. The intervention or procedure presents experiences to subjects that are reasonably commensurate with those inherent in their actual or expected medical, dental, psychological, social or educational situations;
c. The intervention or procedure is likely to yield generalizable knowledge about the subject's disorder or condition which is of vital importance for the understanding or amelioration of the subject's disorder or condition; AND
d. Adequate provisions are made for soliciting assent of the children and permission of the parents or guardians.
4) Research not otherwise approvable which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children if:
a. The IRB finds the above to be true; AND
b. The Secretary of HHS, after consultation with a panel of experts in pertinent disciplines has determined:
1. The research presents a reasonable opportunity to further the understanding, prevention or alleviation of a serious problem affecting the health or welfare of children is present;
2. the research will be conducted in accordance with sound ethical principles;
3. adequate provisions are made for soliciting the assent of children and the permission of their parents or guardians.
NOTE: The regulations define Minimal Risk as the risks of harm anticipated in the proposed research are not greater, considering the probability and magnitude, than ordinarily encountered in the daily life of the research subject, or during the performance of routine physical or psychological examinations or tests. Physical, psychological, social, legal or other risks should be assessed/considered.
Children Who Are in State Custody
Children who are wards of the State or any other agency, institution, or entity may be included in research involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generalizable knowledge about the subject’s disorder or condition, only if such research is:
- Related to their status as ward; or
- Conducted in schools, camps, hospitals, institutions, or similar settings in which the majority of children involved as subjects are not wards.
If the research meets the condition(s) above, an advocate should be appointed for each child who is a ward (one individual may serve as advocate for more than one child), in addition to any other individual acting on behalf of the child as legal guardian or in loco parentis.
The advocate should be an individual who has the background and experience to act in, and agrees to act in, the best interests of the child for the duration of the child’s participation in the research and who is not associated in any way (except in the role as advocate) with the research, the investigators, or the guardian organization. The advocate does not provide informed consent, that is only provided by the legal guardian as determined by DCF.
For guidance on consenting children, see the section on consenting children.
24.4 Non-English Speaking Individuals Participating in Research
Revised 02/01/21
The Department of Health and Human Services (DHHS) regulations for the protection of human subjects require that informed consent information be presented in "language understandable to the subject," and, in most situations, that informed consent be documented in writing (45 CFR §46.116(b)(2) and §46.117(b) (1).
Steps must be taken to assure true informed consent is obtained when non-English speaking individuals are being approached for research participation. This does not simply mean that a form is signed, but rather that steps are taken to assure the study and voluntary nature of the research is understood by the subject. An interperter may need to be involved in the informed consent discussion and a translated consent document may be needed. Further, the IRB may require the investigator to submit a back-translation of the informed consent.
If the majority of subjects are expected to be non-English speaking, use of the translated Long Form Consent and Authorization Process is required.
If the protocol is already approved for English speaking subjects and a non-English speaking subject presents for participation, the Short Form Consent Process and Authorization Process may be used. Typically, this option is used when a single participant is found to be eligible to participate in research and there is no long form consent translation. A witness to the oral presentation is required.
For information regarding the consent process, see section 9.4 on Informed Consent and HIPAA Authorization Process for Non-English Speaking Individuals.
25. Pregnancy Testing in Minor Research Subjects
A minor is defined as a person under the legal age of full responsibility. All persons under 18 years of age are considered minors. Research involving minors requires special consideration on the part of both the research team as well as the IRB. This is particularly so if the study requires pregnancy testing of minor subjects, either to confirm eligibility or as part of routine safety monitoring, for example, before the administration of study drug. Researchers must consider how results of such tests will be handled and to whom they will be disclosed. This plan must be clearly outlined in the protocol as well as in any consent and assent documents.
Pregnancy Testing Requirement
Any protocols that utilize UVM Medical Center resources, would require point-of-care pregnancy testing. UVM Medical Center staff should refer to UVM Medical Center Policies and Procedures Labpoct 110.21 for information about those procedures. There is no pregnancy testing policy for protocols that only utilize UVM resources.
Plan for Disclosure of Pregnancy
If pregnancy is an exclusion criterion and screening procedures to determine eligibility require a pregnancy test, the researcher must determine whether the results of screening tests will be disclosed to the minor subject’s parent(s)/legal guardian or only to the minor subject. There are factors that may necessitate disclosure to parents even for a screening pregnancy test, or that may make reporting to parents after enrollment inappropriate. Researchers may consider both the age and maturity of the minor subjects, as well as any other factors that may impact the reporting decision, such as developmental delays or other relevant physical or mental characteristics of the subject population being studied. For example, if a researcher is enrolling autistic subjects, a positive pregnancy test in this population, regardless of age, would almost certainly need to be disclosed to the parents whether it was a screening test or not. On the other hand, a pregnancy in a 17-year old participating in a study for which parental permission was waived (see section 11.D. Children, Waiver of Consent would not necessarily need to be reported to parents.
Reporting Pregnancy of a Minor to Authorities
Researchers should be aware that there is no age specified at which point the researcher becomes required to report the pregnancy of a minor under state law. Pregnancy of a minor does not necessarily indicate suspected abuse and many other factors may need to be considered. Researchers may consider both the age and maturity of the minor subjects, as well as any other factors that may impact the reporting decision, such as developmental delays or other relevant physical or mental characteristics of the subject population being studied. State child abuse laws governing healthcare providers or persons receiving information about a child who has received healthcare services outline when mandatory reporting is required. See Section 12. Exceptions to Confidentiality.
Language in Consent Form
Regardless of the specifics of the reporting plan, both the parental consent form and the minor’s assent form (they may be the same form depending on the age of the minor subjects) should clearly outline when pregnancy tests will be performed, to whom the results will be disclosed, and whether there may be any exceptions to this. Additionally, an exception to confidentiality statement must be included when it is possible that suspected child abuse or neglect be revealed, requiring mandatory reporting to regulatory authorities.
26. Human Subject Quality Assurance Reviews
Research Protections staff will conduct periodic protocol reviews to ensure that human subject research activities are conducted in accordance with federal regulations, state laws and institutional policies regarding the protection of human subjects in research. Monitoring for compliance and quality is necessary to meet the terms of UVM’s and UVMMC’s Federal wide Assurances.
The IRB is committed to administering a process that is educational for the personnel involved in human subjects research, with the goal of fostering a collegial environment, and thus contributing to a culture of compliance.
Our protocol review program covers a sampling of all studies, both medical and behavioral, that are currently active. Highest priority is given to high-risk protocols, investigator-initiated protocols with no sponsored oversight, protocols that involve the collection of sensitive personal information, and protocols that are approved for the use of a legally authorized representative. However, all types of research, including exempt, expedited, and research overseen by an external IRB, are subject to quality assurance reviews.
The monitoring process may include representatives from other institutional entities such as a Research Navigator from the College of Medicine, or representatives from the University of Vermont Cancer Center (UVMCC) for cancer-related protocols or the Clinical Research Center (CRC) for studies where those resources are being used.
Reviewers
The IRB analysts are best positioned to know and understand the changes over the life of the approved protocol, therefore they will be conducting the review. Others will be included as necessary, i.e., IRB Chair, IRB Assistant Directors, or other IRB members. When appropriate, representative(s) from collaborating units, i.e., CRC or UVMCC.
Notice
The Investigator and their proxy will be given advance notice of the monitoring visit and the protocol that will be reviewed. A representative number of subject research files will be reviewed.
Preparation
Reviewers will go to where the study files are located to conduct the review. Investigators will need to gather all their protocol-specific IRB correspondence and make available all of the subject research files for review. If source documentation is only available in the electronic medical record, the investigator must provide a person to sit with the reviewer to access requested information. The investigator and/or designee should be prepared to answer questions and be able to produce any other information as requested by the reviewer. Copies of documents reviewed during the review may be requested.
Review
The monitoring process includes a review to determine that:
- the protocol on file with the IRB is the protocol being used and being followed;
- all modifications have been submitted to and approved by the IRB and have been implemented;
- the consent form and consent process documentation being used is that which was approved by the IRB;
- the consent forms are appropriately signed and dated;
- adverse event and unanticipated problem reporting guidelines are being followed; and
- eligibility/ineligibility criteria has been met; and
- other items deemed appropriate for review by collaborative departments.
Exit Interview
An exit interview to discuss the findings will be conducted with the principal investigator or his/her designee. After a preliminary report has been reviewed internally, a final report will be forwarded to the principal investigator. Categories of possible monitoring outcomes are as follows:
- Acceptable;
- Additional action required by Investigator (protocol changes, consent modifications, development of a specific process for IRB review to address minor noncompliance findings); or
- Further Committee review required. This outcome typically indicates that multiple deficiencies were identified during the review that requires further deliberation by the Safety Subcommittee or an Ad-Hoc Noncompliance Subcommittee. If the deficiencies are determined to potentially increase risks to subjects, additional reporting to our regulators and sponsors may be required.
26.1 Human Subject Quality Assurance Reviews on External IRB Studies
New 05/10/2023
The UVM IRB may conduct post-approval monitoring in addition to, or in cooperation with, the External IRB for protocols relying on an External IRB. Research Projections Office staff may conduct for-cause audits or routine post-approval monitoring assessments of research ceded to External IRBs as part of the human subject quality assurance review activities described in Section 26, or as directed by the External IRB. Any local consent and/or HIPAA authorization documents used at UVM will name the UVM IRB as an entity that may access participants’ data.
In preparation for monitoring, UVM/UVMHN Investigators will be asked to provide the UVM IRB with current, External IRB-approved protocol and consent documents as well as regulatory determinations made by the External IRB regarding modifications, reportable new information and CAPAs as applicable. Investigators may be asked to facilitate access to the External IRB or Lead Site’s regulatory submission platform for UVM IRB staff. In addition to the review process described in Section 26, regulatory review will confirm that appropriate reliance documentation is in place, that terms of the reliance agreement are being followed, and that the appropriate local consent/HIPAA language is in the current consent document(s).
The results of any for-cause audit or findings that could represent possible serious or continuing noncompliance or unanticipated problems will be shared with the External IRB following terms described in the reliance agreement.
27. Research Non-Compliance Policy
Revised 6/5/23
The Vice President for Research (VPR) has delegated responsibility to the Committees on Human Research, in accordance with the terms of the Federal-Wide Assurance, to investigate and review potential noncompliance; develop and ensure implementation of appropriate corrective actions; and ensure required reporting of serious and/or continuing noncompliance.
Scope
The UVM IRB will review all cases of noncompliance under its purview involving UVM/UVMHN researchers to assess the level of risk of harm, determine whether the research may safely continue, and specify those conditions necessary for the continued protection of human subjects.
If UVM is relying on a different IRB through a reliance agreement, that designated IRB will be responsible for review of these cases. UVM investigators are required to inform the UVM IRB of the outcome of a compliance investigation following requirements under Section 18.1 Reportable New Information on External IRB Studies.
Guiding Principles
Protection of Human Subjects: The University of Vermont and UVMHN are responsible for safeguarding the rights and welfare of human subjects involved in any research activity.
Fairness: The IRB strives to maintain a review that is impartial and honest, free from self-interest, prejudice, or favoritism.
Communication: The committee will communicate with the PI during the review process at points determined to be appropriate by the IRB designee.
Confidentiality: All IRB discussions and documents regarding a situation of noncompliance are considered confidential and privileged. The IRB cannot, however, guarantee complete anonymity to informants or witnesses. Confidentiality will be maintained to the extent possible to protect privacy and prevent retaliation, while still allowing for a full and fair review. Information may be shared, as described above under Required Reporting.
Conflict of Interest: Any IRB member who feels they have a conflicting interest must recuse themselves from reviewing the issue of noncompliance. IRB members who are also listed as key personnel on the protocol(s) will not participate in the review but may be asked for information.
Procedures: In addition to what has been stated within this policy, the Committee will follow all applicable procedures that are outlined within the Committee Operating Procedures document.
General Noncompliance Review
Investigation of noncompliance may include an allegation (unproved assertion) of noncompliance, a self-disclosure of noncompliance, or any other indication that noncompliance may have occurred. The process for the review of potential noncompliance involves initial administrative review, followed by an inquiry/fact finding process if indicated. Once complete, the IRB decides as to whether the noncompliance is serious, continuing, or neither.
Detailed procedures can be found in Section 27.1 Compliance Review Procedures. At any point during the review process, the IRB designee may at their discretion:
• Recommend intervention for the safety of the research subjects.
• Recommend the temporary suspension of research activities.
• Inform, involve, and/or provide salient documents to the PI, members of the research team, the Department Chair, Dean, legal counsel, or Institutional Officials, as appropriate.
• Initiate reporting per federal regulations.
• Initiate a for-cause monitoring visit.
• Recommend immediate corrective actions.
Determinations of the Fully Convened IRB
If the IRB does not find the noncompliance to meet the definition of serious or continuing, the IRB will work with the PI to develop a corrective action plan to prevent recurrence of the issue. If the IRB determines the noncompliance to be serious and/or continuing, a summary of inquiry/findings, including the IRB determination and required corrective actions is provided to the Institutional Official for required reporting.
Institutional Official Review
The Institutional Official (IO) may accept, revise, add to, or reject any or all the IRB’s recommended corrective actions but may not change the IRB’s determinations related to the level of noncompliance or overturn the IRB’s decision to suspend or terminate IRB approval, to require modifications to the approved protocol, or to require more frequent or higher-level IRB review.
Other review
There may be instances where noncompliance rises to the level of academic or research misconduct. Academic or research misconduct charges are processed in accordance with Research Integrity policies.
Appeal to the Institutional Official (IO) of noncompliance actions
Grounds for appeal to the IO are limited to (1) a violation of university rules, regulations, policies; or (2) a specific act by the University that was arbitrary or capricious. Appeals must be filed within 10 business days of receipt of the summary report by the PI. The IO will respond in writing within 30 business days of receiving the appeal, unless, in the opinion of the IO, that is insufficient time to appropriately investigate and consider the substance of the appeal. If additional time is needed, the IO shall contact parties to provide a new date by which the decision shall be made.
Follow-Up
Principal investigators are responsible for ensuring the corrective actions outlined in the final noncompliance report are implemented by the timeframes in the report. Assigned IRB Regulatory Analysts will oversee the progress of implementation with the assistance of the IRB Chair as needed. Failure to meet the conditions established in the report will result in additional review by the IRB and possible termination or suspension of IRB approval.
Notifications
The PI’s Department Chair and the IO are provided a copy of the finalized report. Other UVM offices are informed as necessary to conduct the review or meet reporting requirements. For example, Sponsored Projects Administration may be contacted for advice on requirements for sponsored research related reporting.
External Reporting
When applicable, the IO will report incidents of serious or continuing noncompliance must be reported to the Office of Human Research Protections per the requirements set forth in 45 CFR 46.103(b)(5) and the funding agency or sponsor in accordance with their requirements. Similarly, reports of serious or continuing noncompliance will be provided to the Food and Drug Administration for FDA-regulated research in accordance with 21 CFR 56.108(b), 21 CFR 56.113, 21 CFR 812.150. When appropriate, preliminary reports may be filed pending final resolution of the case. The RPO will redact identifying information from the final summary of findings prior to forwarding final reports to external entities.
27.1. Non-Compliance Review Procedures
Scope
This document describes the procedures for review of all noncompliance matters whether minor, serious or continuing. Relevant definitions and examples are located in section 27.2.
Initial Review of Allegation or Indication of Noncompliance
When there is an allegation or indication of noncompliance, the first step is an administrative review to determine if, in the judgement of the person(s) conducting the review, there is the potential for serious or continuing noncompliance. The initial review may be conducted by the IRB Regulatory Analyst, IRB Director, RPO Director, an IRB Chair or Associate Chair, or another Institutional Representative. Allegations/indications which are determined to have no potential to be serious and/or continuing noncompliance are resolved through informal resolution by the IRB Regulatory Analyst.
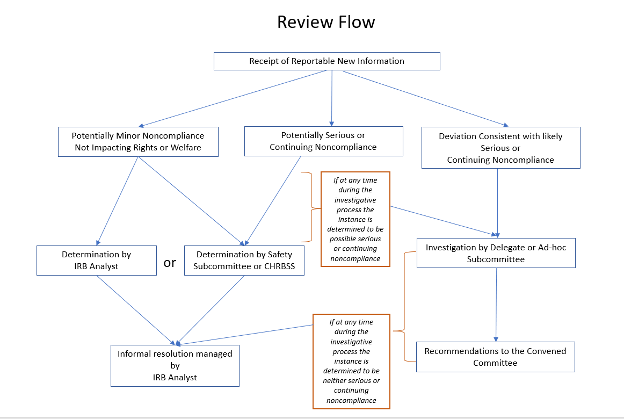
Minor Noncompliance Not Impacting Rights or Welfare - Informal Resolution
The IRB has predetermined that one-time or isolated cases of the following types of incidents constitute minor noncompliance not affecting rights or welfare and can be resolved by IRB Analysts:
• Unapproved changes to personnel, other than the PI or Supervising Investigator, when:
o Any new personnel have completed required human subjects protection training before being involved in human subjects research activities, or
o Removal of personnel does not adversely affect the qualifications of the research team or its ability to safely carry out the research.
• Unapproved changes in the type of compensation (e.g., changing from cash to gift cards), when the change does not alter:
o the amount of compensation; and
o plans for pro-rating compensation.
• Wording changes in recruitment materials or consent forms that do not change the meaning of the information provided or result in excluding any required element(s) of consent;
• privacy or confidentiality provisions (e.g., no changes to the collection of identifiers, whether identifiers are connected to data, etc.).
IRB Analysts assess the totality of the circumstances regarding the incident. If they determine the incident is eligible for informal resolution, it is handled as follows:
• Verify the incident is a one-time or isolated case by checking IRB records. Repeated incidents associated with a PI or project may constitute continuing noncompliance and are not eligible for informal resolution.
• Rectify the issue by advising the PI (and Mentor) to
o promptly obtain IRB approval by submitting an Amendment for Modification if they have not already done so; a reasonable deadline for completing this step should be established if appropriate; and
o adhere to the approved protocol until the Amendment for Modification receives IRB approval (if appropriate).
• Educate the PI (and Mentor) by informing them of the minor noncompliance and reminding them that IRB approval must be obtained for any changes prior to implementation.
• Document the incident as a minor noncompliance event in UVMClick. Documentation supports identification of patterns of noncompliance.
The incident will be forwarded for review in accordance with the Compliance Review Process outlined in the next section if:
• IRB staff determine the incident is not appropriate for informal resolution; or
• The PI does not address the issue within a reasonable timeframe.
Potential Serious or Continuing Noncompliance IRB Review and Resolution
Inquiry/Fact-finding process by delegate or Ad-hoc Subcommittee: If the noncompliance has the potential to be serious or continuing, or if questions remain following the initial review, then an inquiry (fact- finding) process will begin. Fact-finding activities may include, but are not limited to, a for-cause protocol audit by members of the IRB committee, interviews with participants, and meetings with the PI and/or study team. The circumstances of the noncompliance will determine when the fact finding begins and when the full IRB committee is briefed. The PI will be notified of the start of the noncompliance investigation and a discussion will take place regarding whether it would be prudent for the PI to voluntarily place on hold on further accruals while the investigation is ongoing. The fact-finding is delegated by the IRB to the RPO Director, IRB Director, an IRB Chair (Associate Chair or Chair) or an Ad-Hoc Noncompliance sub-committee of the Full. If an Ad-Hoc subcommittee is convened, it will consist of IRB leadership, IRB staff and minimally three additional IRB members whose areas of expertise are suited to the area of study. The IRB may be briefed at any point throughout the fact-finding process, as deemed appropriate by the designee. Investigative review materials including dates of meetings, notes and background information are retained in a dedicated folder within the shared drive for the IRB. Access to these documents is restricted to IRB staff and shared with members to conduct the review.
The fact-finding process continues until the designee or subcommittee has arrived at a recommendation of determination (i.e., serious noncompliance and/or continuing noncompliance, or neither). A draft confidential summary of findings report is then prepared based upon the information gathered. The report includes initial allegation, background information, outcome and recommendations of subcommittee review, outcomes of meeting with PI, and recommended determination and corrective actions. This summary of findings report will be shared with the PI, and if applicable, other person(s) involved at the PI’s request. All parties will be provided an opportunity to respond to any factual inaccuracies within the report before it is sent to the committee for deliberation. The PI will be provided an opportunity to address the IRB at the meeting where the case is scheduled to be discussed.
How does the IRB make a determination: At a convened meeting, the IRB will consider all available information from the summary of findings report and discussion with the PI, as applicable. The members deliberate and vote as to whether the noncompliance is serious noncompliance and/or continuing noncompliance, or neither. The following factors will be taken into consideration by the IRB during their deliberation. As each situation is unique, the indicators of noncompliance that are important in one case may not be relevant in other cases.
Factors in the Determination of Serious Noncompliance:
• Level of risk or potential risk to subjects
• Severity of violation
• Frequency or number of minor deviations or errors
• Intent
• Threat to integrity of the IRB review processes and requirements for the protection of human subjects (i.e., falsification of IRB documents, failure to submit modifications)
• Other factors that, in the judgement of the IRB or designee, are relevant to the situation being reviewed.
Factors in the Determination of Continuing Noncompliance:
• Similarity of noncompliance to previous deviations and/or noncompliance within the same protocol
• Similarity of noncompliance to previous deviations and/or noncompliance in other protocols conducted by the investigator
• Likelihood that instances of noncompliance will continue without intervention.
Final Determination of the IRB: If, in the judgement of the fully convened committee, the noncompliance is neither serious nor continuing, this determination will be shared with the PI. If, in the judgement of the fully convened committee, the noncompliance is serious and/or continuing a final determination will be accompanied by a corrective action plan and be shared with the PI and the Chair of his/her department and others following Section 27.0.
Development of Corrective Action Plans: The subcommittee or designee will develop a proposed plan for corrective actions based on the information gathered during fact-finding and input from the principal investigator and/or other affected individuals. Recommendations may include, but are not limited to:
• Additional human subjects’ protection training for the PI/research team;
• The investigator and/or other affected individuals develop and submit a corrective action plan;
• Recommendation to the Institutional Official (or designee) to work with the PI’s department chair/head to determine appropriate actions regarding the data collected as a result of the non-compliant actions*
• Review of internal departmental or institutional mechanisms and systems for opportunities to prevent recurrence or similar occurrences by others;
• Require additional oversight/mentorship (e.g., by other faculty member or department process);
• Require more frequent IRB reviews;
• Require internal monitoring visits or monitoring plans;
• Suspend or terminate individual protocols;
• Restrict researcher’s research activities.
*Only the Institutional Official has the authority to restrict use of research data. Therefore, the Institutional Official will be consulted prior to the fully convened meeting if there are initial recommendations to restrict use of the research data. Examples of restrictions include but are not limited to full restriction on uses of the research data or biospecimens, prohibition on publication, restrictions on release of data to a regulatory agency for consideration of approval of a test article, request to destroy data or biospecimens.
27.2. Definitions, Guidance, and Examples Referenced as Part of Non-Compliance Reviews
Noncompliance: Conduct of research in a manner that deviates from the approved protocol or disregards or violates federal regulations or institutional policies and procedures applicable to human subjects’ research. Noncompliance may result from actions or omissions by study personnel and can range from relatively minor or technical deviations to serious deviations that threaten subjects’ rights or welfare.
Noncompliance can result from action or omission, and may be minor, serious, and/or continuing as defined below.
Minor noncompliance: Noncompliance that does not rise to the level of serious or continuing noncompliance. Minor noncompliance typically involves administrative oversights, non-substantive unapproved changes, etc.
Noncompliance that does, or reasonably may, adversely affect the rights, safety, or welfare of research subjects is not minor, even if no actual harm has occurred.
Examples that may be deemed minor include, but are not limited to:
• Implementing non-substantive changes to approved procedures without IRB approval, such as:
o Wording changes in recruitment materials or consent forms that do not change the meaning of the information provided or result in excluding any required element(s) of consent;
o Changing the order in which study conditions are administered, as long as a specific order is not necessary to minimize risk;
• Enrolling subjects who do not meet the inclusion or exclusion criteria, when the safety and/or wellbeing of the participant is not compromised.
• Unapproved changes in study personnel, when the changes do not alter the qualifications of the overall research team and all personnel have completed required human subjects protection training.
• The correct consent version is used to consent a participant but is missing the IRB stamp of approval.
• Research visits out of window
Serious noncompliance: Noncompliance that, in the judgement of the IRB, potentially substantially increases the risk of harm to research subjects and/or reduces benefits to human subjects or compromises the integrity of the research oversight process. The IRB does not have to find that harm has occurred, or was likely to occur, to make a determination of serious noncompliance.
Acts that are determined by the IRB to be flagrant or intentional violations of IRB requirements may also constitute serious noncompliance. The IRB will consider the circumstances surrounding the case when making a decision related to serious noncompliance.
In general, examples of serious noncompliance may include, but are not limited to:
• Allowing unqualified or untrained individuals to perform research procedures or monitor subject safety;
• Failure to obtain written informed consent when the safety and/or wellbeing of the participant is potentially compromised.
• Failure to provide participants with all information necessary to constitute meaningful initial or ongoing informed consent.
• Enrolling a child in a research study without the written informed consent of a parent or legal guardian (DCF)
• Enrolling subjects from a vulnerable population (i.e., children, prisoners, cognitively impaired individuals, subordinates, etc.) when their inclusion is not described in the IRB-approved protocol and appropriate protections are not in place;
• Enrolling subjects who do not meet the approved eligibility criteria when doing so compromises the safety or well-being of the subjects;
• Failure to follow approved measures for protecting privacy and confidentiality when the failure presents any risk of harm to the research subject (such as harm to their reputation, social or psychological harm, risks of legal or civil liability, embarrassment, harm to workplace or family relationships, etc.);
• Implementing unapproved changes to research activities that increase risks to participants or adversely affect their rights, safety, or welfare (e.g., adding survey questions that collect sensitive information, substantially increasing the duration or intensity of exercise activities, adding plans to collect data from private records without subject consent, changes to confidentiality protections, etc.);
• Failure to report serious adverse events or unanticipated problems involving risks to subjects or others as required by IRB policy;
• Providing false or intentionally misleading information to the IRB;
• Multiple issues suggesting a lack of oversight, inaction, or negligence
Continuing noncompliance: Noncompliance defined as a pattern of noncompliance (recurring or ongoing) that in the judgement of the IRB, may indicate any of the following:
• an underlying deficiency in knowledge of the regulations or IRB requirements
• an unwillingness or inability to comply with the regulations that protect the rights and welfare of participants and others
• compromises the scientific integrity of a study such that important conclusions can no longer be reached
• suggests a likelihood that non-compliance will continue without intervention
• involves frequent instances of minor noncompliance
• failure to respond to inquiries from the IRB to resolve an open case of non-compliance or pattern of minor non-compliance.
Examples of continuing noncompliance may include, but are not limited to:
• Repeated failure to obtain IRB approval prior to initiating human subjects research activities;
• Continuing to engage in noncompliant activities after being notified or advised of concerns;
• Recurring lapses in approval;
• Multiple instances of serious or minor noncompliance; this includes multiple incidents within a single project or multiple incidents by a single investigator across more than one project when the safety and/or wellbeing of the participant is potentially compromised.
• Failure to respond to incidents of noncompliance or failure to enact required corrective actions.
Noncompliance in Exempt research: Research granted exempt status is subject to fewer regulatory requirements than non-exempt research. For example, some changes to an exempted protocol can be implemented without prior IRB review. In general, noncompliance in exempt research involves the following:
o Initiating human subjects research activities prior to receiving a written determination of exemption from the IRB.
o Deviations from an exempted protocol that alter the exempt status of the study.
27.3 Non-compliance Policy on External IRB Studies
UVM/UVMHN Investigators are required to notify the UVM IRB of an External IRB’s intended investigation of possible serious or continuing noncompliance according to Section 18.1 of the Policies and Procedures. In most cases, in accordance with the reliance agreement that is in place, the External IRB will take the lead on the review and investigation of serious or continuing noncompliance. The UVM IRB may be requested by the External IRB to conduct an investigation on their behalf, or the investigative process may be shared between the two IRBs. The UVM IRB will follow the Noncompliance Procedures located in Section 27.1 of the Policies and Procedures as well as terms set forth in the reliance agreement.
Under the reliance agreement the UVM IRB will have the opportunity to review corrective and preventive action plans proposed by the External IRB, prior to local implementation, and reserves the right to enact a higher level of CAPA.
The External IRB is responsible for reporting to applicable regulators and sponsors. The External IRB must provide the UVM IRB adequate opportunity to review their findings prior to reporting, in accordance with the reliance agreement. The UVM IRB will report to the Institutional Officials and other stakeholders as applicable.
28. Electronic Signatures Policy
Background
Since August 1997, 21 CFR Part 11 has established the requirements for electronic records. It also determined standards to make electronic signatures trustworthy, reliable, and essentially equivalent to paper records and handwritten signatures. It was created primarily to prevent fraud while permitting the widest possible use of electronic technology to reduce costs of paper processes.
UVMClick
UVM/UVMMC utilize Huron’s Click electronic research administration software. Huron’s Click Portal Compliance Extranet product meets or exceeds the requirements in 21 CFR Part 11 for “closed” systems by providing manageable name/password administration, strong passwords with rotation options and electronic signature components that are only executable by, and designed to be used only by, specified individuals for specific approval actions. The IRB has CLICK’s 21 CFR Part 11 Compliance statement on file.

29. Regulatory Definitions
Revised 10/5/2023
o Absent. Absent for discussion and voting for reasons other than a conflict of interest.
o Abstain. Present for the vote, but not voting “for” or “against”.
o Accrual. The number of subjects who have completed or are actively in the process of completing a study. This does not include screen failures. It does include withdrawals. Example: You enroll 100 to accrue 25.
o Administrative hold. A voluntary action initiated by the principal investigator whenever specific research activities, including subject enrollment, are placed temporarily on hold.
o Adverse event. An untoward or undesirable experience associated with the use of a medical product, such as a drug, device, or biologic, in a patient or research subject.
o Advocate. An individual who has the background and experience to act in, and agrees to act in, the best interest of the child for the duration of the child's participation in the clinical investigation.
o Agent. For purposes of this document, an institution's employees or agents refers to individuals who: (1) act on behalf of the institution; (2) exercise institutional authority or responsibility; or (3) perform institutionally designated activities. "Employees and agents" can include staff, students, contractors, and volunteers, among others, regardless of whether the individual is receiving compensation. A student's affiliation with an academic institution makes him or her an agent of that institution; and thus, the academic institution is engaged in the research regardless of where the research takes place.
o Allegation of noncompliance. An unproven assertion of noncompliance.
o Alternate member. Alternate Institutional Review Board (IRB) committee members may be designated, as needed, for regular voting members. The appointment of alternate members should be based on expertise similar to that of the regular voting member. An alternate member may vote only when the regular voting member is absent.
o Anonymous data. Information that was previously recorded or collected without any of the 18 identifiers as defined by HIPAA, and no code is assigned that would allow data to be traced to an individual.
o Assent. A child's affirmative agreement to participate in research. Mere failure to object should not, absent affirmative agreement, be construed as assent.
o Assured institution. An institution with a federal wide assurance (FWA) that has filed with the federal Office for Human Research Protections. Employees and agents of the institution holding an approved FWA are covered whenever they are involved in the conduct of the research covered by the FWA. Employees and agents are individuals performing institutionally designated activities and acting on behalf of the institution or exercising institutional authority or responsibility.
o Belmont Report. Report by the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research identifying the basic ethical principles underlying the conduct of research involving human subjects, that is, respect for persons, beneficence, and justice.
o Beneficence. Persons are treated in an ethical manner not only by respecting their decisions and protecting them from harm, but also by making efforts to secure their well-being. Such treatment falls under the principle of beneficence. Two general rules have been formulated as expressions of beneficent actions (Belmont Report, 1978):
1. Do no harm, and
2. Maximize possible benefits and minimize possible harms.
o Benign behavioral interventions. Interventions brief in duration, harmless, painless, not physically invasive, not likely to have a significant adverse lasting impact on subjects, and the investigator has no reason to think the subjects will find the interventions offensive or embarrassing. Examples:
o Requesting subjects play an online game
o Solve puzzles under various noise conditions
o Decide how to allocate a nominal amount of received cash between themselves and someone else.
o Biologic product. A biological product (biologic) is a medical product. Many biologics are made from a variety of natural sources, such as humans, animals, or microorganisms. Like drugs, some biologics are intended to treat diseases and medical conditions. Other biologics are used to prevent or diagnose diseases. Examples of biological products include:
1. Vaccines
2. Blood and blood products for transfusion and or manufacturing into other products
3. Allergenic extracts, which are used for both diagnosis and treatment, such as allergy shots
4. Human cells and tissues used for transplantation, such as tendons, ligaments, and bone
5. Gene therapies
6. Cellular therapies
7. Tests to screen potential blood donors for infectious agents, such as HIV
8. In general, the term "drugs" includes therapeutic biological products
o Broad Consent. Broad consent is an option for storage, maintenance, and secondary research use of identifiable private information and identifiable biospecimens. Not in use at UVM.
o Certification. Official notification by the institution to the supporting Federal department or agency component, that a research project or activity involving human subjects has been reviewed and approved by an IRB in accordance with an approved assurance.
o Children. People who have not attained the legal age for consent to treatments or procedures involved in the research, under the applicable law of the jurisdiction in which the research will be conducted.
o Clinical investigation. The Food and Drug Administration (FDA) has defined clinical investigation to be synonymous with research. The FDA defines clinical investigation as any experiment that involves a test article and one or more human subjects, and that either must meet the requirements for prior submission to the FDA or the results of which are intended to be later submitted to, or held for inspection by, the FDA as part of an application for a research or marketing permit.
o Clinical Trial. Clinical trial means a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of the interventions on biomedical or behavioral health-related outcomes.
o Coded Data Set. Identifying information that would enable the investigator to readily ascertain the identity of the individual to whom the private information or specimens pertain has been replaced with a number, letter, symbol, or combination thereof and a key to decipher the code exists that would enable linkage of the identifying information to the data or biospecimens. Coded data sets are not considered “de-identified” when the “code” is the study subject number.
o Conflict of interest. Any interest that could reasonably be expected to affect the objectivity of an IRB member or consultant in relation to an application or other matter under IRB review. An IRB member or consultant has a conflict of interest if the individual:
1. Is or will be an investigator or member of the research team (that is, listed on the IRB application)
2. Has an immediate family member (that is, spouse, dependent children) or personal relationship with an individual who is one of the investigators
3. Has a financial or managerial interest in a sponsoring entity or product being evaluated or provided by a commercial entity in the research, as defined by UVM/UVMMC policies
4. Has received or will receive compensation with value (as defined by UVM/UVMMC policies) that may be affected by the outcome of the research project
5. Has a proprietary interest in the research, such as a nonprovisional patent application, patent, trademark, copyright, or licensing agreement as defined by UVM/UVMMC policies
6. Has a nonfinancial interest (personal circumstance, ethical belief, or other factor) that may be conflicting, for example, the IRB member has an interest that he or she believes conflicts with his or her ability to review a project objectively
7. Has responsibility for institutional business development, such as raising funds or garnering support for research or as an officer within the Department of Development
o Consent capacity. An individual's ability to understand and process information relevant to making an informed, voluntary decision to participate in research. Several kinds of information are crucial to such decisions, including an understanding of the purpose of the study, its experimental nature, risks and anticipated benefits, the right to withdraw, alternatives to participation, confidentiality protections, and the safeguards used to minimize risks. A wide variety of diseases, disorders, conditions, situations, and injuries can affect a person's ability to understand such information, to weigh the advantages and disadvantages of participation in research, and to reach an informed decision regarding study participation.
o Consent document. A structured, written description in understandable terms of relevant research project information. The consent document is not consent itself; it is the record of what has been communicated to a potential participant. It is the document that ensures all regulatory elements are present and communicated to a potential participant. When signed by the potential participant, the consent document is a record of the receipt of research-related information by the participant. It also serves as reference material for the participant as the research project progresses. It is not a contract and is not legally binding, and the participant may choose to withdraw consent at any time.
o Consultant. A scientist or nonscientist from within or external to UVM/UVMMC who has special expertise to act — at the request of the IRB — as an ad hoc reviewer of a research project application. These individuals have access to all documents relevant to the specific project under review, may participate in the deliberations and make recommendations on the project, but may not vote and are not counted toward quorum.
o Continuing noncompliance. A pattern of noncompliance (recurring or ongoing) that in the judgement of the IRB, may indicate any of the following:
o an underlying deficiency in knowledge of the regulations or IRB requirements
o an unwillingness or inability to comply with the regulations that protect the rights and welfare of participants and others
o compromises the scientific integrity of a study such that important conclusions can no longer be reached
o suggests a likelihood that non-compliance will continue without intervention
o involves frequent instances of minor noncompliance
o failure to respond to inquiries from the IRB to resolve an open case of non-compliance or pattern of minor non-compliance.
o Continuing review. Periodic review of research activities at intervals appropriate to the degree of risk, but not less than once per year. The criteria for approval are defined by federal regulations. Unless the IRB determines otherwise, continuing review will no longer be required for: projects under expedited categories, projects requiring limited IRB review, or projects that have progressed to data analysis (including analysis of identifiable private information or identifiable biospecimens).
o Cooperative research project. Research projects that involve more than one institution as defined by federal regulations.
o Coordinating center. An institution, department or center that agrees to be responsible for the conduct and administrative or coordinating functions of a multicenter research project.
o Co-principal investigator (co-PI). UVM/UVMMC investigator who plays a key role in scientific development and conduct of the study. The co-PI collaborates with the principal investigator who has overall responsibility for study conduct. Conditions of eligibility for the role of co-PI are the same as for a PI.
o Covered entity. HIPAA regulations apply to health plans, health care clearinghouses and health care providers who transmit health information. Any individual creating or accessing protected health information (PHI) for the delivery of health care at UVM/UVMMC is within the covered entity.
o Custom device. Custom device means a device that:
1. Necessarily deviates from devices generally available or from an applicable performance standard or premarket approval requirement in order to comply with the order of an individual physician or dentist.
2. Is not generally available to, or generally used by, other physicians or dentists.
3. Is not generally available in finished form for purchase or for dispensing upon prescription.
4. Is not offered for commercial distribution through labeling or advertising; and
5. Is intended for use by an individual patient named in the order of a physician or dentist and is to be made in a specific form for that patient or is intended to meet the special needs of the physician or dentist in the course of professional practice (such as a particular operating tool).
o Data Custodian. Individuals who will have actual possession of the data files or biospecimens, and who will be responsible for observance of all conditions of use, including the establishment and maintenance of security arrangements to prevent unauthorized use.
o Data Set. A dataset is a structured collection of data generally associated with a unique body of work.
o Data safety monitoring board (DSMB). A data safety monitoring board is an independent committee set up specifically to monitor data throughout the duration of a study to determine if continuation of the study is appropriate scientifically and ethically. Factors that suggest a DSMB is needed:
1. A large study population and
2. Multiple study sites. It is more difficult to recognize a pattern of increased or unusual problems or events when investigators treat small fractions of the population separately.
3. Highly toxic therapies or dangerous procedures.
4. High expected rates of morbidity or mortality in the study population.
5. High chance of early termination of the study. DSMB membership is usually comprised of experts in the fields of medicine and science that are applicable to the study — statistical experts, lay representatives and others who can offer an unbiased assessment of the study progress.
o Data safety monitoring plan (DSMP). A DSMP is a quality-assurance plan for a research study. A data and safety monitoring plan (DSMP) is meant to ensure that each clinical investigation has a system for appropriate oversight and monitoring of the conduct of the clinical investigation. The purpose of a DSMP is to ensure the safety of the participants, the validity of the data and the integrity of the study, and the appropriate termination of studies for which significant benefits or risk has been uncovered or when it appears that the investigation cannot be concluded successfully. A DSMP is commensurate with the risks involved with the research study. The DSMP may include a data and safety monitoring board (DSMB).
o Data use agreement. An agreement into which UVM/UVMMC and the investigator enter with the intended recipient of a limited data set that establishes the ways in which the information in the limited data set may be used and how it will be protected.
o Dead fetus. A fetus that exhibits neither heartbeat, spontaneous respiratory activity, spontaneous movement of voluntary muscles nor pulsation of the umbilical cord.
o Deception. The action of intentionally misleading or providing incomplete disclosure to a research participant for research purposes. Providing that the research is otherwise minimal harm or harmless, the IRB will approve deception research as long as it has a sound debriefing process for the participant.
o De-identified health information. Health information that has been stripped of all 18 identifiers, related to the patient and the patient’s relatives, employers, and household members), as defined by the Health Insurance Portability and Accountability Act of 1996 and its implementing regulations (HIPAA), the releasing entity has no actual knowledge that the information could be used alone or in combination with other information to identify an individual who is a subject of the information. Importantly, in small populations (including small states such as Vermont), characters Data sets may also be de-identified within the meaning of HIPAA using an “expert determination,” however this method is unusual in the context of research.
o De-identified health information is not protected by HIPAA, and therefore is not subject to its regulations. However, UVM/UVMHN policy may still require appropriate data sharing agreements.
The 18 HIPAA identifiers
1. Names
2. All geographic subdivisions smaller than a state*
3. Telephone numbers
4. Fax numbers
5. Email Addresses
6. Social Security numbers
7. Medical record numbers
8. Health plan beneficiary numbers
9. Account numbers
10. Certificate/license numbers
11. Vehicle identifiers and serial numbers, including license plate numbers
12. Device identifiers and serial numbers
13. Web Universal Resource Locators (URLs)
14. Internet Protocol (IP) address numbers
15. Biometric identifiers, including finger and voice prints
16. Full face photographic images and any comparable images
17. All elements of dates (except year)**
18. Any other unique identifying number, characteristic or code
*including street address, city, county, precinct, zip code, and their equivalent geocodes, except for the initial three digits of a zip code, if according to the current publicly available data from the Bureau of the Census: (1) The geographic unit formed by combining all zip codes with the same three initial digits contains more than 20,000 people; and (2) The initial three digits of a zip code for all such geographic units containing 20,000 or fewer people is changed to 000.
**for dates directly related to an individual, including birth date, admission date, discharge date, date of death; and all ages over 89 and all elements of dates (including year) indicative of such age, except that such ages and elements may be aggregated into a single category of age 90 or older.
o Delivery. Complete separation of the fetus from the woman by expulsion or extraction or any other means.
o Direct identifiers - An identifier that links to one specific person, can be used by itself to identify the person (e.g., name, social security number, medical record number, medical device number, email address, etc.).
o Disclosure of PHI. The release, transfer, or provision of access to, or divulging in any manner of, information outside the covered entity.
o Documentation. The act or an instance of furnishing or authenticating with documents.
o Emergency research. Planned research involving human subjects who have a life-threatening medical condition that necessitates urgent intervention (for which available treatments are unproven or unsatisfactory), and who, because of their condition (such as traumatic brain injury) cannot provide informed consent.
o Emergency treatment IDE. A mechanism through the FDA for providing eligible participants with investigational devices for the treatment of an immediate serious or life-threatening illness for which there are no satisfactory alternatives.
o Emergency treatment IND. A mechanism through the FDA for providing eligible participants with investigational drugs, agents, or biologics for the treatment of an immediate serious or life-threatening illness for which there are no satisfactory alternatives.
o Emergency use. Use of a test article on a human subject in a life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval [21 CFR 56.102(d)].
1. Life-threatening includes both life-threatening and severely debilitating diseases or conditions where likelihood of death is high unless the course of the disease is interrupted, and diseases or conditions with potentially fatal outcomes, where the endpoint of clinical trial analysis is survival.
2. The criteria for life-threatening do not require the condition to be immediately life-threatening or to immediately result in death. Rather, the subjects must be in a life-threatening situation requiring intervention before review at a convened IRB meeting is feasible.
3. Severely debilitating: Diseases or conditions that cause major irreversible morbidity, such as blindness, loss of arm, leg, hand or foot, loss of hearing, paralysis, or stroke.
o Engagement of institutions in human subject research. An organization is considered engaged in human research when its employees or agents, for the purposes of the nonexempt research project, obtain:
1. Data about the subjects of the research through intervention or interaction with them.
2. Identifiable private information about the subjects of the research.
3. The informed consent of human subjects for the research; or
4. When the institution receives a direct federal award to conduct human subject research, even when all activities involving human subjects are carried out by a subcontractor (that is, employees or agents of another institution).
o Enrollment. Occurs when an eligible, informed, potential participant undergoes the initial informed consent process and voluntarily agrees to participate in a research project. Example: You enroll 100 to accrue 25. See also Accrual.
o Exempt human subjects research. Studies determined by the IRB to meet the exempt criteria as defined by the federal regulations. Exempt studies do not require periodic review by the IRB unless a change in the project is planned.
o Expedited review. A review of research involving human subjects by the IRB chair or by one or more experienced reviewers designated by the chair from among members of the IRB in accordance with the requirements set forth in 45 CFR 46.110.
o Experimental subject (as defined by Department of Defense, or DOD). Research involving a human being as an experimental subject is an activity, for research purposes, where there is an intervention or interaction with a human being for the primary purpose of obtaining data regarding the effect of the intervention or interaction [32 CFR.210.102 (f) reference (c)]. Examples include, but are not limited to, a physical procedure, a drug, a manipulation of the subject or subject's environment, and the withholding of an intervention that would have been undertaken if not for the research purpose.
o Expired study. When continuing review of the research does not occur prior to the end of the approval period specified by the IRB, IRB approval expires automatically. The study expires on the date specified on the approval letter and the consent document. No activities can occur after the expiration date.
o External Unanticipated Problem. A problem or event involving research subjects enrolled by other institutions in multicenter research projects that fall under the purview of an external IRB, that is, not the UVM IRB.
o Federal wide assurance (FWA). A formal, written, binding attestation in which an institution ensures to the Department of Health and Human Services (HHS) that it will comply with applicable regulations governing research with human subjects.
o Fetus. The product of conception from implantation until delivery.
o Final report. A report the principal investigator may elect to submit to the IRB to serve as a final record of any pertinent activity since the last continuing review report and to record research project completion.
o Fluctuating capacity. Capacity to consent may alter as a function of the natural course of an illness, response to treatment, effects of medication, general physical health, and other factors. Therefore, a participant's ability to provide ongoing informed consent must be re-evaluated periodically throughout the course of his or her participation in a study.
o Food and Drug Administration (FDA). The regulatory authority in the United States that oversees the pharmaceutical and medical device industries. The FDA is responsible for ensuring that the drugs and medical devices marketed in the U.S. are safe and have a greater benefit than risk when used according to manufacturer's directions.
o Full committee review. Studies reviewed by the full, convened IRB committee with a recorded vote and corresponding minutes to document the discussion.
o Generalizable Knowledge. Generalizable knowledge includes one or more of the following concepts: (1) The information contributes to a theoretical framework or an established body of knowledge; (2) The primary beneficiaries of the study are other researchers, scholars, and practitioners in the field of study; (3) Publication, presentation or other distribution of the results is intended to inform the field of study; and (4) The results are intended to be replicated in other settings
o Greater than minimal risk. The research involves more than minimal risk to subjects.
o Guardian. An individual who is authorized under applicable state or local law to consent on behalf of a child to general medical care when general medical care includes participation in research.
o HIPAA authorization. A customized document or form that gives permission to use specified protected health information (PHI) for a specific purpose, or to disclose PHI to a third party specified by the investigator other than for treatment, payment, or health care operations.
o HIPAA Privacy Rule. The HIPAA Privacy Rule for the conduct of research (45 CFR 164.501) establishes the conditions under which protected health information may be used or disclosed by covered entities for research purposes. Refer to HHS.gov for additional information regarding the conduct of research.
o Honest Broker. An honest broker is an individual who has access to the desired data by virtue of his or her responsibilities and who is not involved as a listed researcher on the respective research study.
o The honest broker accesses the desired medical record information and provides the researcher with de-identified data or a limited data set.
o If the honest broker is providing the researcher with a limited data set, the broker must present an internal data use agreement to the researcher prior to receiving the data set.
o The honest broker can assign a code to the data, provided that the researcher does not have access to the information linking the code to the identities of the research subjects. Using the code, the researcher can request, through the honest broker, additional medical information corresponding to a given research subject.
o If the honest broker provides coded data to the research but not the method to de-code the data, then the information provided will be considered de-identified or a limited data set depending upon the data elements included in the data set.
o Human biospecimens. A quantity of tissue, blood, urine, or other human-derived material. A single biopsy may generate several biospecimens, including multiple paraffin blocks or frozen biospecimens. The molecular makeup of such specimens reflects the physiologic or pathologic condition of the person from whom they derive; therefore, they provide sensitive and specific insight into the biologic state of the donor. A biospecimen can include subcellular structures (such as DNA), cells, tissue (such as bone, muscle, connective tissue, and skin), organs (such as liver, bladder, heart, and kidney), blood, gametes, embryos, fetal tissue, and waste (such as urine and stool). Portions or aliquots of a biospecimen are referred to as samples. (Derived from National Cancer Institute Best Practices for Biospecimen Research.)
o Humanitarian use device exemption (HDE). An FDA approval for a physician to use a humanitarian use device (HUD) in clinical treatment or as the subject of a clinical investigation.
o Humanitarian use device (HUD). A device that is intended to benefit patients by treating or diagnosing a disease or condition that affects fewer than 4,000 individuals in the U.S. per year.
o Human specimen research repository. A collection of human specimens and associated data for research purposes, the physical structure where the collection is stored, and all relevant processes and procedures.
o Human subject as defined by Department of Defense:
1. Research involving a human being as an experimental subject. An activity, for research purposes, where there is an intervention or interaction with a living individual for the primary purpose of obtaining data regarding the effect of the intervention or interaction.
2. Research involving human subjects. An activity that includes both a systematic investigation designed to develop or contribute to generalizable knowledge and involve a living individual about whom an investigator conducting research obtains data through intervention or interaction with the individual or identifiable private information.
o Human subject as defined by FDA. An individual who is or becomes a subject in research, either as a recipient of the test article or as a control. A subject may be either a healthy human or a patient. A human subject includes an individual on whose specimen a medical device is used.
1. Test article: Any drug (including a biological product for human use), medical device for human use, human food additive, color additive, electronic product, or any other article subject to FDA regulation.
Human subject as defined by HHS. Human subject means a living individual about whom an investigator (whether professional or student) conducting research:
Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens, OR
Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
o Human subject identifier. Any word, number, symbol, or combination of words, numbers or symbols that can be used by a third party to uniquely identify an individual, such as name, Social Security number, address or patient registration number that is provided for use in a research protocol.
o Identifiable private information. Is private information for which the identity of the subject is or may readily be ascertained by the investigator or associated with the information.
o Identifiable biospecimen. A biospecimen for which the identity of the subject is or may readily be ascertained by the investigator or associated with the biospecimen.
o Impaired consent capacity. Impaired consent capacity may involve partial impairment, impairment that fluctuates over time or complete impairment. For example, consent capacity can be affected by a wide range of disorders and conditions, such as dementia, stroke, traumatic brain injury, developmental disorders, serious mental illness, intoxication, and delirium.
o Impartial Witness. is a person who is independent of the trial and cannot be unduly influenced by the people involved with the trial and who attends the informed consent process if the participant or the participant's LAR cannot read and who reads the ICF and any other written information supplied to the participant. They may also be asked to sign the consent form. This definition contains four parts, all of which must be met. Here they are presented separately for emphasis and analysis:
1. “Who is independent of the trial:” This could be a person who is a family member. It would not be a member of the site staff involved with the study.
2. “Who cannot be unfairly influenced by people involved with the trial:” This would be a person free from potential coercion or undue influence or conflicted interest.
3. “Who attends the informed consent process if the subject or the subject’s legally acceptable representative cannot read:” This emphasizes the participation of the witness throughout the consent process, not just when the subject signs. A robust informed consent process will likely result, on the part of the person obtaining consent.
4. “Who reads the informed consent form and any other written information that was supplied to the subject:” This responsibility has the witness confirming the subject was presented sufficient information to assure truly informed consent of the subject.
o Indirect identifiers. An identifier that does not link to one specific person but can be used in combination with other information to identify a person (e.g., dates including dates of birth, dates of death, zip codes, cities, counties, etc.).
o Individually identifiable health information. Any information collected from an individual (including demographics) that is created or received by a health care provider, health plan, employer, and or health care clearinghouse that relates to the past, present or future physical or mental health or condition of an individual, or the provision of health care to an individual or the past, present or future payment for the provision of health care to an individual and identifies the individual and or to which there is reasonable basis to believe that the information can be used to identify the individual.
o Informed consent. An ongoing process of communication between the participant and the study team. Informed consent is a continuing process by which a participant, after having been informed, voluntarily confirms his or her willingness to participate in a research project and can demonstrate understanding of all aspects of the research project that are relevant to the participant's decision to participate.
o Institutional official. The institutional official (IO) who is the signatory on the federal wide assurance (FWA) filed with OHRP to ensure compliance with regulations governing protection of human subjects. OHRP requires the institutional official to be a high-level official who has the authority to represent the institution named in the FWA.
o Institutional review board (IRB). A specifically constituted review body established or designated by an entity to protect the rights and welfare of human subjects recruited to participate in biomedical or behavioral or social science research.
o Interaction. Includes communication or interpersonal contact between investigator and subject.
o Intervention. Physical procedures by which information or biospecimens are gathered, such as venipuncture, and manipulations of the subject or the subject's environment that are performed for research purposes.
o Investigational agent. A pharmaceutical form of an active ingredient or placebo being tested or used as a reference in a clinical trial. This includes products with a marketing authorization when used or assembled (formulated or packaged) in a way different from the approved form, products used for an unapproved indication or products used to gain further information about an approved use.
o Investigational device. Any health care product that does not achieve its primary intended purposes by chemical action or by being metabolized. A medical device that is the subject of a clinical study designed to evaluate the effectiveness and or safety of the device. Investigational use also includes clinical evaluation of certain modifications or new intended uses of legally marketed devices.
o Investigational device exemption (IDE). Application document submitted to the FDA proposing human clinical research to study an unapproved significant risk device, or a cleared or approved device for use other than its approved indication or intent. FDA grants permission so a device that otherwise would be required to comply with a performance standard or to have premarket approval can be shipped lawfully for the purpose of conducting investigations of that device. This FDA permission is evidenced by the assignment of an IDE number.
o Investigational drugs or investigational biologics. New drugs or biologics that have not yet been approved by the FDA or approved drugs that have not yet been approved for a new use and are in the process of being tested for safety and effectiveness.
o IND (investigational new drug) application. Means by which permission may be obtained to 1) ship an investigational drug, biologic, or agent across state lines and 2) use in humans prior to FDA review of clinical data that has determined a new drug, agent, or biologic to be safe and effective for a specific use. Testing of an investigational product may proceed once a valid IND is in effect or an IND exemption has been granted.
o IRB authorization agreement. A formal, written agreement in which the reviewing IRB agrees to serve as the IRB of record for a relying institution, including an academic institution. Agreements are generally used to cover a single research study, categories of research studies or research studies within a research program.
o IRB of record. A reviewing IRB that assumes IRB responsibilities for another institution and is designated to do so through an approved federal wide assurance on file with the federal Office for Human Research Protections.
o Key information. Five elements at the beginning of the consent form, and informed consent process, would encompass the required key information. (1) consent is being sought for research and that participation is voluntary (2) the purpose of the research, expected duration, and procedures (3) reasonably foreseeable risk or discomforts (4) benefits to subjects or others that may be reasonably expected (5) alternative procedures or courses of treatment that might be advantageous.
o Label. The FDA-approved label is the official description of a drug or biologic product that includes indication (what the product is used for); who should take it; adverse events (side effects); instructions for uses in pregnancy, children, and other populations; and safety information for the patient. Labels are often found inside product packaging.
o Legally authorized representative (LAR). Legally authorized representative (LAR) means an individual or judicial or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research. If there is no applicable law addressing this issue, legally authorized representative means an individual recognized by institutional policy as acceptable for providing consent in the non-research context on behalf of the prospective subject to the subject’s participation in the procedure(s) involved in the research.
o Legally effective informed consent. A potential participant has been provided enough information to make a decision; the potential participant has the capacity to make a decision; the potential participant understands the consequences of his or her decision; and the potential participant can communicate that decision.
o Limited data set. Protected health information that excludes direct identifiers of individuals, and their relatives, employers, or household members, including:
Direct Identifiers that Must be Removed from Limited Data Sets
1. Names
2. Postal address information, other than town, or city, state, and zip code
3. Telephone Numbers
4. Fax Numbers
5. Email address
6. Social Security numbers
7. Medical Record numbers
8. Health plan beneficiary numbers
9. Account numbers
10. Certificate/license numbers
11. Vehicle identifiers & serial numbers, license plate numbers
12. Device identifiers and serial numbers
13. Web Universal Resource Locators (URLs)
14. Internet Protocol (IP) address numbers
15. Biometric identifiers, including finger and voice prints
16. Full face photographic images and any comparable images
o Limited data sets contain indirect identifiers and therefore are not considered to be de-identified. Specifically, limited data sets may include dates more specific than the year and geographic information including town, city, state, and zip code. Research relying on data from a limited data sets does not require IRB review and approval. However, the process for creating the limited data set, may be considered human subjects research and require IRB review.
o Limited IRB Review. A limited IRB review is making and documenting that there are adequate privacy safeguards for identifiable private information and identifiable biospecimens in research for exempt 2(iii) and 3(i)(c) projects that collect or use sensitive and identifiable data. An exempt determination is issued once the expediting reviewer confirms that these protections are acceptable.
o Local research context. Knowledge of the institution and community environment in which human subjects research will be conducted.
o Material transfer agreement (MTA). A contract that governs the transfer of tangible research materials between two organizations when the recipient intends to use the materials for his or her own research purposes.
o Medical device. A medical device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part or accessory that is:
1. Listed in the online FDA database.
2. Intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
3. Intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes [21 U.S.C. 321(h)].
o Minimal risk. The probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
o Minimal risk for prisoners. The probability and magnitude of physical or psychological harm that is normally encountered in the daily lives or in the routine medical, dental, or psychological examination of healthy persons. The regulations further state that the IRB must find that the risks involved in the research are commensurate with risks that would be accepted by nonprisoner volunteers [45 CFR 46.305(a)(3)]. If Subpart C does not apply, the IRB may use an equivalent definition of minimal risk for prisoners. [45 CFR 46.303(d)]
o Modification. Any change to an IRB-approved study protocol regardless of the level of review it receives initially.
o Neonate. A newborn zero to 28 days old.
o Nonaffiliated member. Any IRB member who is not currently affiliated with UVM/UVMMC and whose immediate family members are not affiliated with UVM/UVMMC. Examples of UVM/UVMMC affiliation include employment, participation as a student in a UVM academic program or receipt of post-employment benefits, such as health and wellness or a pension.
o Noncompliance. Conduct of research in a manner that deviates from the approved protocol or disregards or violates federal regulations or institutional policies and procedures applicable to human subjects’ research. Noncompliance may result from actions or omissions by study personnel and can range from relatively minor or technical deviations to serious deviations that threaten subjects’ rights or welfare.
o Non-unanticipated problem involving risk to subjects or others. A reportable event that does not meet the UVM IRB's definition of an unanticipated event involving risk to subjects or others. See Unanticipated problem involving risk to subjects or others.
o Nonviable neonate. A neonate after delivery that, although living, is not viable.
o Notification. Process of notifying research subjects of changes in the research by letter or phone.
o Not Research. The following activities are deemed not to be research:
(1) Scholarly and journalistic activities (e.g., oral history, journalism, biography, literary criticism, legal research, and historical scholarship), including the collection and use of information, that focus directly on the specific individuals about whom the information is collected.
(2) Public health surveillance activities, including the collection and testing of information or biospecimens, conducted, supported, requested, ordered, required, or authorized by a public health authority. Such activities are limited to those necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance (including trends, signals, risk factors, patterns in diseases, or increases in injuries from using consumer products). Such activities include those associated with providing timely situational awareness and priority setting during the course of an event or crisis that threatens public health (including natural or man-made disasters).
(3) Collection and analysis of information, biospecimens, or records by or for a criminal justice agency for activities authorized by law or court order solely for criminal justice or criminal investigative purposes.
(4) Authorized operational activities (as determined by each agency) in support of intelligence, homeland security, defense, or other national security missions.
o Office for Human Research Protections (OHRP). The office under the Department of Health and Human Services responsible for implementing HHS regulations (45 CFR 46) governing biomedical and behavioral and or social science research involving human subjects.
o Oral (verbal) consent. A spoken presentation of the elements of informed consent to the prospective subject or their legally authorized representative. The presentation may be based on information contained within an oral consent script or the written consent document. Oral consent is often associated with waiving the documentation of consent. Oral consent is usually recorded in the research project files.
o Payment Card Industry Data (PCI) – Includes cardholder data that may appear in the form of the full PAN (Primary Account Number) plus any of the following: cardholder name, expiration date and/or service code. PCI may also include Sensitive Authentication Data that is security-related information including but not limited to card validation codes/values, full track data from the magnetic stripe or equivalent on a chip, PINs, and PIN blocks used to authenticate cardholders and/or authorize payment card transactions.
o Permission. The agreement of parents or guardians to the participation of their child or ward in research.
o Practicably. The word practicable appears in the consent alterations and waiver section but is intentionally left undefined. In the Final Rule preamble, SACHRP’s recommendation is that “this requirement be interpreted to mean that it would be impracticable to perform the research, not impracticable to obtain consent due to financial or administrative burdens, without the waiver or alteration. “
o Pregnancy. Encompasses the period of time from implantation until delivery. A woman shall be assumed to be pregnant if she exhibits any of the pertinent presumptive signs of pregnancy, such as a missed menses, until the results of a pregnancy test are negative or until delivery.
o Preparatory to research. Any action taken in assessing the research question or hypothesis, such as accessing medical records, querying of databases for any type of individually identifiable health information, or any activity where PHI is accessed to prepare a research protocol.
o Principal investigator (PI). Adheres to federal regulations, state and local laws, institutional policies, IRB policies and procedures regarding the safety and protection of human subjects, and good clinical practice (GCP) guidelines. The principal investigator ensures adherence by:
1. Supervising the research process.
2. Taking responsibility for ensuring that key study personnel are properly trained, qualified, and have appropriate facilities and resources to conduct the research.
3. Ensuring adherence to the study protocol.
4. Monitoring the informed consent process.
5. Communicating regularly and effectively with the research staff.
6. Taking responsibility for protecting the safety and welfare of research subjects.
o Prisoner. Any individual involuntarily confined or detained in a penal institution. The term is intended to encompass individuals sentenced to such an institution under a criminal or civil statute, individuals detained in other facilities by virtue of statutes or commitment procedures that provide alternatives to criminal prosecution or incarceration in a penal institution, and individuals detained pending arraignment, trial or sentencing. If Subpart C does not apply, the IRB may use an equivalent definition of prisoners. [45 CFR 46.303(c)]
o Prisoner of war. Any person captured, detained, held or otherwise under the control of Department of Defense personnel (military and civilian, or contractor employee) except DOD personnel held for law enforcement purposes (DOD directive 3216.2, section 4.42).
o Privacy versus confidentiality. Privacy is about people and their choice to share personal information. It is a right in health care and research. Confidentiality is about data. It is the investigator's obligation to protect subjects' information.
o Private information. Information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place. It includes information that has been provided for specific purposes by an individual, and that individual can reasonably expect will not be made public, such as a medical record.
o Protected health information. Individually identifiable health information, regardless of format, that is collected by a Covered Entity and relates to the past, present, or future physical or mental condition of a patient, the provision of healthcare or past, present, or future payment for the provision of healthcare. Protected health information can include demographic information (such as names, email addresses, telephone numbers, etc.) as well as information relevant to a person’s health such as dates of disease onset, testing, treatment, etc., a particularly rare health condition, rash, birthmark, or any other information that could be used to identify the patient (or members of the patient’s family, employer and others who live in the patient’s household. Protected health information excludes individually identifiable health information in (i) Education records covered by the Family Educational Rights and Privacy Act (FERPA); (ii) Records described at 20 U.S.C. 1232g(a)(4)(B)(iv); and Employment records held by a covered entity in its role as employer; however, those records are covered by other privacy laws and requirements. Additionally, data generated by a Part 2 entity (federally assisted entities that hold themselves out as providing and do provide substance use disorder treatment) are protected by heightened privacy rules set forth in separate regulations. The University of Vermont Health Network has two Part 2 programs—UVMMC’s Addiction Treatment Program and UVMMC’S Day One Program.
o Personally Identifiable Information (PII). Any information about an individual maintained by an agency, including (1) any information that can be used to distinguish or trace an individual ‘s identity, such as name, social security number, date and place of birth, mother ‘s maiden name, or biometric records; and (2) any other information that is linked or linkable to an individual, such as medical, educational, financial, and employment information.
o Protocol deviation. Any divergence or departure from the expected conduct of an IRB-approved study that is not consistent with the current, approved research protocol, consent process or document or study modification.
o Protocol violation. Problems that violate the terms of a study but do not meet the criteria for an unanticipated risk to subjects or others. See Unanticipated problem involving risk to subjects or others.
o Public Health Authority. Agency or authority that is responsible for public health matters as part of its official mandate.
o Reconsenting. Process of notifying research subjects of changes in the research, including documentation of the subject's continued informed consent through signature on a revised written consent form.
o Recruitment. Recruitment, a component of the consent process, is the process of distributing or presenting information that describes the research project and eligibility criteria so that a prospective subject may consider enrollment.
o Recruitment screening/waivers. Allows waiver of informed consent for subject recruitment or screening, under certain conditions.
o Recusal. Absent from the meeting during discussion and voting because of a conflict of interest.
o Relying organization. An organization, including an academic institution, with whom UVM has either entered into an IRB Authorization Agreement, or an agreement entered into as part of a cooperative research project.
o Reportable event. A process (with an associated IRB form) used by an investigator to report any problem or event or other act or omission to the IRB that in their opinion is an unanticipated problem involving risk to subjects or others.
o Repository. A repository compiles data, specimens, or both for future research purposes. The repository receives, processes, stores, and distributes data with or without specimens to researchers. The repository may or may not have an honest broker.
o Research activities. Research activity includes all contact with the research subject (such as enrolling subjects, intervention, or interaction), data collection and data analysis.
o Research (as defined by DOD). An activity that includes both a systematic investigation designed to develop or contribute to generalizable knowledge and involves a living individual about whom an investigator conducting research obtains data through intervention or interaction with the individual or identifiable private information, or activities covered by section 32 CFR 219.101 (including exempt research involving human subjects) and DOD Instruction 3216.02.
o Research (as defined by FDA). Any experiment that involves a test article and one or more human subjects, and that meets any one of the following:
1. Must meet the requirements for prior submission to the FDA under section 505(i) of the Federal Food, Drug, and Cosmetic Act, meaning any use of a drug other than the use of an approved drug in the course of medical practice
2. Must meet the requirements for prior submission to the Food and Drug Administration under section 520(g) of the Federal Food, Drug, and Cosmetic Act, meaning any activity that evaluates the safety or effectiveness of a device
3. Any activity the results of which are intended to be later submitted to, or held for inspection by, the FDA as part of an application for a research or marketing permit
o Research (as defined by HHS). A systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge. Activities that meet this definition constitute research for purposes of this policy, whether or not they are conducted or supported under a program that is considered research for other purposes. Some demonstration and service programs may include research activities. For the purposes of this part, the following activities are deemed not to be research:
1. Scholarly and journalistic activities (e.g., oral history, journalism, biography, literary criticism, legal research, and historical scholarship), including the collection and use of information, that focus directly on the specific individuals about whom the information is collected.
2. Public health surveillance activities, including the collection and testing of information or biospecimens, conducted, supported, requested, ordered, required, or authorized by a public health authority. Such activities are limited to those necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance (including trends, signals, risk factors, patterns in diseases, or increases in injuries from using consumer products). Such activities include those associated with providing timely situational awareness and priority setting during the course of an event or crisis that threatens public health (including natural or man-made disasters).
3. Collection and analysis of information, biospecimens, or records by or for a criminal justice agency for activities authorized by law or court order solely for criminal justice or criminal investigative purposes.
4. Authorized operational activities (as determined by each agency) in support of intelligence, homeland security, defense, or other national security missions.
o Research involving a human being as an experimental subject (as defined by DOD). An activity, for research purposes, where there is an intervention or interaction with a living individual for the primary purpose of obtaining data regarding the effect of the intervention or interaction. Research involving a human being as an experimental subject is a subset of research involving human subjects. This definition relates only to the application of section 980 of Title 10 USC; it does not affect the application of 32 CFR 219. This definition does not include activities that are not considered research involving human subjects, activities that meet the exemption criteria at section 32 CFR 219.101(b), and research involving the collection or study of existing data, documents, records, or specimens from living individuals. Section 980 of Title 10 USC imposes limitations on waiving informed consent when using DOD appropriated funds. Section 980 of Title 10 USC is applicable only to DOD funded research involving a human being as an experimental subject. Section 980 of Title 10 USC is not applicable to exempt research involving human subjects.
o Research involving human subjects (as defined by DOD). An activity that includes both a systematic investigation designed to develop or contribute to generalizable knowledge and involves a living individual about whom an investigator conducting research obtains data through intervention or interaction with the individual or identifiable private information, or activities covered by section 32 CFR 219.101 (including exempt research involving human subjects) and DOD Instruction 3216.02.
o Secondary research use. Re-using for research purposes identifiable and non-identifiable information and biospecimens that are collected for some other primary or initial activity (such as, health records, archives, from research studies other than the proposed research study).
o Screen failures. Subjects who consented to participate in research but who were disqualified during screening procedures.
o Sensitive Information - Information, the loss, misuse, or unauthorized access to or modification of, that could adversely affect the national interest or the conduct of federal programs, or the privacy to which individuals are entitled under 5 U.S.C. Section 552a (the Privacy Act), but that has not been specifically authorized under criteria established by an Executive Order or an Act of Congress to be kept classified in the interest of national defense or foreign policy.
o Serious noncompliance. Any noncompliance that, in the judgement of the IRB, potentially substantially increases the risk of harm to research subjects and/or reduces benefits to human subjects or compromises the integrity of the research oversight process. The IRB does not have to find that harm has occurred, or was likely to occur, to make a determination of serious noncompliance.
o Short-form consent document. A written consent document stating the elements of consent have been presented orally to a non-English speaker. A witness to the oral presentation is required.
o Significant risk (SR) device study. A study of a device that presents a potential for serious risk to the health, safety, or welfare of a participant and:
1. Is intended as an implant.
2. Is used in supporting or sustaining human life, or otherwise prevents impairment of human health.
3. Is of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise prevents impairment of human health; or
4. Otherwise presents a potential for serious risk to the health, safety, or welfare of a participant.
o Single IRB. Single IRBs have overall regulatory review responsibilities (by agreement) for multiple-site research protocols. U.S. Institutions engaged in cooperative (multi-site) research must rely upon approval by a single IRB by January 2020.
o Sponsor-investigator. An individual who both initiates and conducts an investigation, and under whose immediate direction:
1. The investigational drug is administered or dispensed, and or
2. The investigational device is administered, dispensed, or used. The term does not include any person other than an individual.
o Study expiration. If IRB approval of a specific study expires before continuing review and approval occur, investigators must stop all research activities involving human subjects related to that study except where they judge that it is in the best interests of already enrolled subjects to continue to participate. When investigators make this judgment, they must promptly notify the IRB. When the IRB reviews the investigator's decision, it may decide whether it is in the best interests of already-enrolled subjects to continue to participate in the research by considering the best interests of subjects either one at a time or as a group. If an IRB determines that it is not in the best interests of already-enrolled subjects to continue to participate, investigators must stop all human subjects research activities, including intervening or interacting with subjects, or obtaining or analyzing identifiable private information about human subjects. Investigators may resume the human subjects research activity once continuing review and approval by the IRB has occurred.
o Substitution. Alternate members of the Committee are allowed to substitute their vote in the absence of a regular member for whom they have the same level of expertise (e.g., alternate member M.D. can substitute for regular member M.D.)
o Surrogate consent. Consent obtained from the participant's legally authorized representative (LAR).
o Suspension for cause. An action initiated by the IRB to stop temporarily some or all research procedures pending future action by the IRB or by the investigator or his or her personnel. Examples of a suspension for cause might include:
1. Inappropriate involvement of human subjects in research
2. Violation of the rights or welfare of human subjects or others
3. Serious or continuing noncompliance with federal regulations or IRB policies
4. New information regarding increased risk to human subjects or others
o Termination for cause. An action initiated by the IRB to stop permanently some or all research procedures.
o Test article. Any investigational drug, biologic product, such as blood or a vaccine, or medical device for human use.
o Therapeutic misconception. The term "therapeutic misconception" is used to describe the assumption by research participants that decisions about their care are being made solely with their benefit in mind. Therapeutic misconception can be defined as the situation where a participant or a LAR either overestimates the direct therapeutic benefits that may be gained by participation in the research and/or underestimates the risks, thereby compromising his or her ability to provide and/or maintain a voluntary and knowing informed consent. Investigators must be especially careful to make participants and their families or caretakers aware of the differences between individualized treatment versus research and the separate and distinct roles of the clinician and the research investigator.
o Treatment investigational device exemption (IDE). A mechanism through the FDA for providing eligible participants with investigational devices for the treatment of a serious or life-threatening illness for which there are no satisfactory alternatives.
o Treatment investigational new drug (IND). A mechanism through the FDA for providing eligible participants with investigational drugs for the treatment of a serious or life-threatening illness for which there are no satisfactory alternatives.
o Unanticipated problem involving risk to subjects or others. Any unanticipated problem or adverse event that meets these three criteria:
1. Serious. Serious problems or events that result in significant harm (which may be physical, psychological, financial, social, economic, or legal) or increased risk for the subject or others (including individuals who are not research subjects). These include:
1. Death.
2. Life-threatening adverse experience.
3. Hospitalization, whether inpatient, new or prolonged.
4. Disability and or incapacity, whether persistent or significant.
5. Birth defect or anomaly.
6. Breach of confidentiality; and
7. Other problems, events, or new information (such as publications, DSMB reports, interim findings, product-labeling change) that in the opinion of the local investigator may adversely affect the rights, safety or welfare of the subjects or others, or substantially compromise the research data.
2. Unanticipated. Unexpected problems or events are those that are not already described as potential risks in the protocol consent document, not listed in the investigator's brochure or not part of an underlying disease. A problem or event is unanticipated when it was unforeseeable at the time of its occurrence. A problem or event is unanticipated when it occurs at an increased frequency or at an increased severity than expected.
3. Related. A problem or event is related if it is possibly related to the research procedures.
o Viable. As it pertains to the neonate, means being able, after delivery, to survive (given the benefit of available medical therapy) to the point of independently maintaining heartbeat and respiration.
o Vulnerable populations in research. Vulnerable populations are individuals that are vulnerable to coercion or undue influence, such as children, prisoners, or individuals with impaired decision-making capacity, or economically or educationally disadvantaged persons.
o Ward. A child who is placed in the legal custody of the state or other agency, institution, or entity, consistent with applicable federal, state, or local law.
o Withdrawals. Subjects who signed the consent form, but later withdrew from the study, either before or after receiving a study drug, device, or intervention. This does not include screen failures.
o Written or in Writing. Writing on a tangible medium (e.g., paper) or in an electronic format.

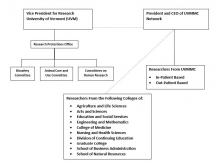{kind=link}
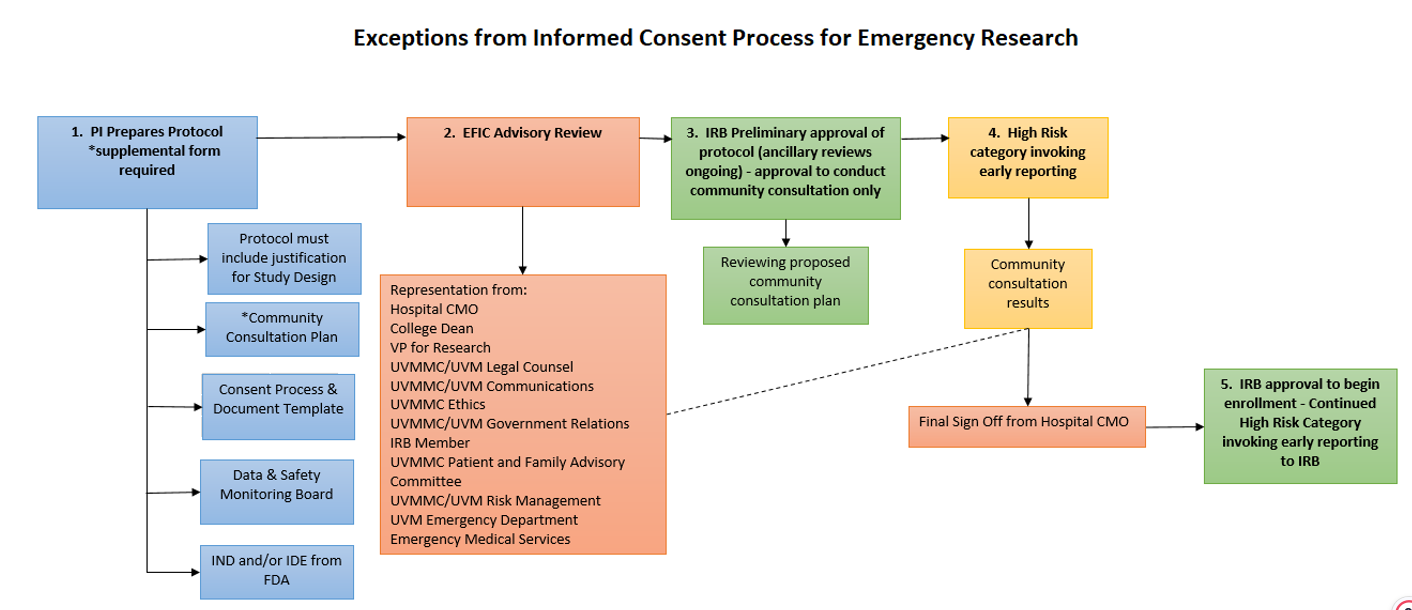{kind=link}
{kind=link}
31. 2018 Common Rule Transition
The final revisions to the Common Rule (Federal Policy for the Protection of Human Subjects) became effective on January 20, 2020. Accordingly, the IRB implemented the following changes.
Policies and procedures may continue to change as a result of evolving interpretations and guidance so be sure to check the website for updates.
- Past exempt submissions remained under the Pre-2018 Rule. Effective January 21, 2019, new exemptions will be processed under the New Rule.
- New studies processed after January 21, 2019 will be processed under the New Rule.
- Existing studies were reviewed at time of continuing review and determinations made as to whether they should be transitioned. Those protocols meeting specific criteria to cease annual reviews were identified and documented in the system. Those protocols with increased risk, FDA-regulated and/or continued enrollment, were amended to enable transition to the New Rule.
- New cooperative research protocols processed after January 20, 2020 are in compliance with the New Rule (falling under single IRB). UVM will rely but not act as the reviewing IRB. WIRB will assist those researchers wishing to be the lead site for cooperative research studies. Existing cooperative research studies will remain under the Pre-2018 Rule thus not requiring IRB reliance.
Updated 12/16/2022